
Церебральная и спинальная атаксия что это такое
Описание
Атаксия спиноцеребеллярная представляет собой совокупность генетических недугов, которые носят неврологический характер, проявляются нарушением деятельности базальных ядер головного мозга и мозжечка и передаются по наследству. В результате этого изменяется координация движений и прочее. Негативным в заболевании является тот факт, что на сегодняшний день какого-либо определенного лечения не существует. Болезнь наследуется по механизму аутосомной доминантности, когда ферменты, которые появляются в результате мутаций генов структурных белков, деформируются. Обычно наследование происходит по отцовской линии из поколения в поколение.
Этиология
Поскольку спиноцеребеллярная атаксия является болезнью наследственной, то она встречается в каждом последующем поколении, где был больной отец. Заболевание само по себе не слишком распространено (от одного до двадцати четырех заболевших на сто тысяч человек). При этом разные типы недуга встречаются в разных странах мира. В современной медицине существует больше двадцати вариантов этой болезни.
Классификация
Спиноцеребеллярная атаксия (мкб 10 - G11) в 90% случаев представляет собой шесть из двадцати генетических вариантов болезни. Данные варианты были классифицированы по номерам: 1, 2, 3, 6, 7 и 8 типы атаксии. Патология выражается в изменении количества CAG в части генов, которые кодируют больные гены. Рассмотрим далее эти типы подробнее:
- Атаксия первого типа сегодня самая распространенная. Она возникает вследствие размещения в шестой хромосоме мутированного гена ATXN1. В норме этот ген имеет тридцать шесть повторов, при большем же их количестве развивается болезнь. Мутация гена вызывает образование ДНК белка, который принимает участие в метаболизме клеток. Это способствует дегенерации и развитию болезни.
- СА второго типа распространена несколько меньше. Она характеризуется увеличением повторов в двенадцатой хромосоме. Какую функцию при этом выполняет белок, медицине неизвестно.
- Третий тип именуется болезнью Мачадо-Джозефа. В этом случае нарушение происходит в гене, что размещен в четырнадцатой хромосоме. Белок при этом принимает участие в обмене энергией между мозжечком и ядрами мозга.
- Спиноцеребеллярная атаксия шестого типа является редким недугом. Здесь происходит нарушение в гене, который находится в девятнадцатой хромосоме. Ген кодирует белок, который размещается в нейронах мозжечка. Этот процесс вызывает также наследственную форму мигрени.
- СА седьмого типа обуславливается нарушениями гена в третьей хромосоме. Какую функцию выполняет белок, медицине неизвестно.
- Восьмой тип характеризуется изменениями гена в тринадцатой хромосоме.
Причины

При любом типе заболевания происходит мутация гена, приводящая к образованию ДНК белка патологической формы, который богат глутамином. Он вызывает появление в ядрах нейронов мозжечка и базальных ядер мозга отложений в виде агрегатов, нарушая свойства протеинов. Белки принимают участие в обмене веществ, протекающих в нервной ткани. Скорость протекания данного процесса зависит от количества поворотов в гене, которое отличается от нормы. Это определяет симптоматику заболевания. При созревании половых клеток симптомы усиливаются.
Симптомы
Все типы данного заболевания имеют одинаковую симптоматику, различными могут быть только второстепенные элементы. Так, спиноцеребеллярной атаксии симптомы не проявляются в детском возрасте. Средний возраст людей, страдающих недугом, составляет от восемнадцати до тридцати лет. Атаксия третьего, шестого и седьмого типов развивается позже, обычно это происходит после тридцати лет. Первым признаком недуга является появление неуклюжести при ходьбе и беге. Позже наблюдается тремор конечностей, изменение походки, офтальмоплегия, почерк меняется. Со временем недуг приводит к развитию паркинсонизма. При некоторых видах болезни наблюдается атрофия зрительного нерва. СА шестого, седьмого и восьмого типов характеризуется нарушением речи и процесса глотания, что приводит к истощению. Истощение вместе с патологиями часто провоцируют смертельный исход. При всех видах заболевания нарушается координация движений. Средняя продолжительность жизни больных составляет от десяти до двадцати пяти лет, в зависимости от формы недуга и качества ухода за ними.

Диагностика
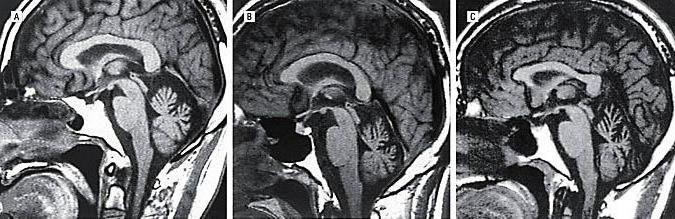
Прежде всего, проводят неврологический осмотр больного, изучают анамнез, проводят МРТ и различные молекулярно-генетические анализы и ДНК -диагностики . На разных стадиях развития недуга врач выявляет разные нарушения, связанные с неврологией. Это может быть тремор, нарушение речи, дисфагия и прочее. Некоторые формы заболевания обуславливаются быстрым развитием зрительных нарушений, что приводят к полной слепоте. Заболевание склонно прогрессировать. При исследовании наследственного анамнеза может быть обнаружено аутосомно-доминантное наследование от отца. МРТ показывает нарушения в области больших полушарий, мозжечка и базальных ядер. Может также наблюдаться атрофия мозжечка. Молекулярное исследование и ДНК- диагностика указывают на увеличенное число повторов в генах больного.
Лечение
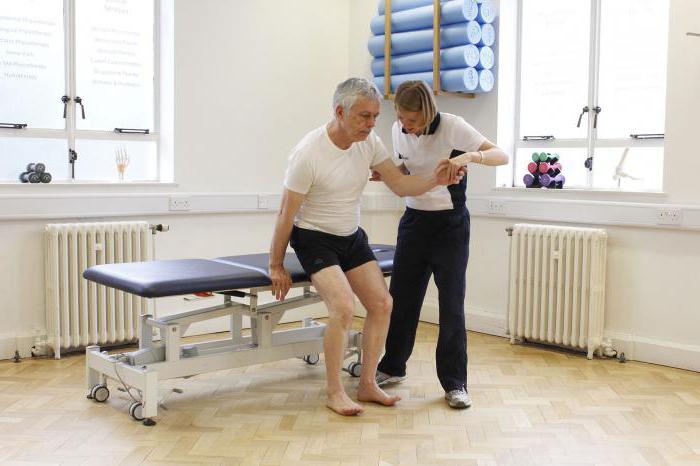
Какого-либо эффективного лечения данного недуга на сегодняшний день нет. Результативность поддерживающей терапии на данный момент не доказана, однако она проводится для замедления развития недуга. Так, спиноцеребеллярная атаксия лечение предполагает в виде витаминотерапии и средств, которые стимулируют обмен веществ и метаболизм в нервной ткани. Также больным назначают ноотропные препараты. Немаловажную роль играет и физическая культура. Врачи рекомендуют больным выполнять комплекс упражнений для укрепления мышц и уменьшения нарушений равновесия. Проводят сеансы массажа и электромиостимуляцию.
Прогноз
Как правило, прогноз при данном заболевании неблагоприятный, поскольку недуг постоянно прогрессирует и приводит к инвалидизации, а затем и к летальному исходу. Поэтому ответ на вопрос о том, лечится ли спиноцеребеллярная атаксия, будет отрицательным. Данное наследственное заболевание неизлечимо. В некоторых случаях прогноз может быть не столь негативным. Это бывает при развитии заболевания в преклонном возрасте и своевременном лечении, тогда большое количество симптомов могут не проявиться. Если недуг обнаружился в молодом возрасте, длительность жизни таких пациентов будет невелика. Больные с пятым и шестым типом недуга живут нормальной жизнью немного дольше, обычно срок их жизни не меняется. При правильном уходе и своевременной терапии врачам удается увеличить время жизни больных на десять лет. В среднем с таким заболеванием, как спиноцеребеллярная атаксия, живут около двадцати лет. Причиной летального исхода часто становится сердечная недостаточность и наличие инфекций.
Профилактика
Профилактические меры представляют собой медицинское и генетическое консультирование родителей, в чьем анамнезе наблюдались такие состояния. Также проводится генетическая перинатальная диагностика. Врачом определяется риск появления недуга у прямых родственников. Риск развития патологии для здоровых братьев и сестер, а также детей больного составляет 50%. В свою очередь, дети этих людей имеют вероятность унаследовать болезнь в 25%. Все эти лица находятся в группе риска и представляют собой главные объекты для консультирования. Основой профилактического исследования является ДНК-диагностика лиц из группы риска на наличие мутированных генов.
Спиноцеребеллярная атаксия в современное время является тем заболеванием, которое не лечится и приводит со временем к летальному исходу. Предупредить развитие заболевания можно при помощи специальных методов диагностики, но предотвратить его развитие невозможно, поскольку недуг этот имеет наследственный характер и обуславливается мутациями здоровых генов. Все это подталкивает современную медицину к разработке методов исследования на самых ранних этапах развития болезни, а также изучению причин мутаций, которые передаются по наследству от отца к детям.

Ежедневно человек выполняет большое количество целенаправленных движений, в реализации которых участвуют разные мышечные группы (мышцы-антагонисты, мышцы-синергисты, мышцы-агонисты, мышцы-фиксаторы), вестибулярный аппарат, головной мозг, спинной мозг, зрительный анализатор.
Одним из самых сложных двигательных процессов в нашем организме считается ходьба и поддержание равновесия. Мышцы туловища и конечностей последовательно сокращаются и расслабляются – появляется шаг, центральная нервная система регулирует скорость шага, инициирует и прекращает ходьбу, адаптирует движение к изменяющимся условиям внешней среды (подъем, спуск и др.), зрительный анализатор передает информацию об окружающем мире, вестибулярный аппарат участвует в поддержании равновесия в пространстве.
Атаксия может быть статической, когда человек не может удержать равновесие в стоячем положении, или динамической — нарушается координация движений.
В зависимости от того, в каком образовании расположен очаг поражения, выделяют несколько видов атаксий:
- вестибулярная;
- корковая;
- мозжечковая;
- сенситивная и др.
Спиноцеребеллярная дегенерация
Термин спиноцеребеллярная дегенерация используется для обозначения группы заболеваний нервной системы, которые проявляются нарушением координации движений в результате сочетанного поражения мозжечка, ствола головного мозга и спинного мозга. В эту группу входят идиопатические и наследственные спиноцеребеллярные атаксии.
Для всех атаксий данной группы общим является прогрессирующее расстройство движений и сочетанное поражение структур ЦНС.
Среди всех наследственных заболеваний ЦНС атаксии занимают по частоте второе место после нервно-мышечных заболеваний. На сегодняшний день единая классификация данной группы не разработана, так как наследственные атаксии по клиническим проявлениям очень разнообразны и порой не ясно, это один и тот же вариант или другой.
Остановимся на самых распространенных формах наследственных спиноцеребеллярных дегенераций.
Каждый 120 человек в мире является носителем мутантного гена, который отвечает за развитие болезни. Заболевание передается по аутосомно-рецессивному типу одинаково часто мальчикам и девочкам.
Причины
Механизм развития атаксии связан с нарушением синтеза на митохондриях белка фратаксина, который регулирует транспорт ионов железа через клеточные мембраны. В результате мутации происходит гибель митохондрий и клеток ЦНС, поджелудочной железы, миокарда и других органов. В ЦНС повреждаются спинно-мозжечковые пути, задние корешки спинного мозга, задние и боковые столбы спинного мозга. На поздних стадиях болезни в патологический процесс вовлекаются ядра черепных нервов, ножки мозжечка, зубчатое ядро, периферические нервы. Все перечисленные структуры отвечают за координацию движений.
Симптомы
Первые клинические симптомы появляются у детей в возрасте старше 10 лет. Ребенок начинает испытывать трудности при движении в темноте, появляется неуверенная походка, спотыкания и пошатывания. Постепенно нарушается звукопроизношение (дизартрия), координация движений в руках, которая проявляется затруднением в письме и изменением почерка. По мере прогрессирования заболевания появляется слабость и атрофия (гибель) мышц ног и рук, которые приводят к полной утрате самостоятельного передвижения и приковывают человека к постели. Тазовые расстройства в виде недержания мочи и кала выявляются у большинства пациентов. У некоторых больных происходит атрофия зрительных нервов, проявляющаяся полной или частичной утратой зрения. Слабоумие может быть не у всех. На поздних стадиях болезни человек теряет способность к самообслуживанию.
При осмотре у невролога выявляется важный симптом атаксии Фридрейха — исчезновение сухожильных рефлексов (коленные, ахилловы и др.) вплоть до полной арефлексии.
В патологический процесс вовлекается сердце, опорно-двигательный аппарат, эндокринная система. Человек предъявляет жалобы на боли в области сердца, одышку, сердцебиение. Развивается кардиомиопатия. Симптомы со стороны сердечно-сосудистой системы могут появиться намного раньше неврологических проявлений.
Из эндокринных нарушений диагностируют сахарный диабет, дисфункцию яичников, нарушение полового созревания, ожирение и другие заболевания.
Продолжительность жизни при атаксии Фридрайха редко составляет больше 20 лет. Основная причина летального исхода – развитие сердечной или легочной недостаточности, инфекционных осложнений.
Выделяют атипичную форму атаксии, которая характеризуется более поздним дебютом (30–50 лет), благоприятным течением, медленным прогрессированием. При этой форме не выявляют кардиомиопатию, эндокринные нарушения, угасание рефлексов.
Диагностика
- ДНК-тестирование считается решающим методом в диагностике атаксии Фридрейха.
- ЭНМГ (электронейромиография) является одним из важных методов исследования, при котором выявляют поражение чувствительных спинномозговых путей при сохранности двигательных нервов.
- МРТ. Диагностируют атрофию (гибель) вещества спинного мозга.
- ЭХО-КГ, ЭКГ и другие исследования обнаруживают изменения со стороны сердечной мышцы.
- Анализ крови на сахар, гормональный профиль диагностирует эндокринные изменения.
- Рентгенография позвоночника, стоп помогает выявить отклонения.
Диагноз ставится на основании данных всех методов исследования.
Лечение
Специальная терапия не разработана. Все терапевтические мероприятия носят симптоматический характер.
- Лекарственные препараты (никотиновая кислота, рибофлавин, аскорбиновая кислота) улучшают работу митохондрий.
- Физиолечение (электростимуляция).
- Массаж и ЛФК.
- Ортопедические мероприятия: стельки и обувь, операции на позвоночнике.
Причины
В основе заболевания лежит мутация гена, который отвечает за встраивание витамина Е в структуру липопротеидов низкой плотности (участвуют в антиоксидантной защите клеток).
Симптомы
По клиническим проявлениям атаксию с дефицитом витамина Е практически невозможно отличить от атаксии Фридрейха. Первые симптомы в виде нарушения координации движений появляются у детей в возрасте от 4 лет до 18 лет. Нарушение речи, угасание рефлексов также встречаются при этой форме. Следует отметить, что поражение сердечно-сосудистой, костной и эндокринной системы встречается в несколько раз реже. Ближе к 30 годам большинство пациентов теряют способности к самостоятельному передвижению и самообслуживанию, приковываются к постели.
Диагностика
Всем пациентам с атаксиями необходимо проводить анализ крови на определение сывороточного витамина Е. Снижение или отсутствие его концентрации в сыворотке крови является достоверным маркером заболевания.
Лечение
Назначение пожизненной терапии витамином Е в суточной дозировке 5-10 мг\кг приводит к исчезновению симптомов и клиническому выздоровлению человека, особенно при раннем начале лечения.
Эта группа атаксий включает в себя ряд самостоятельных заболеваний, которые очень трудно отличить друг от друга по клиническим проявлениям.
Развитие генной инженерии позволило классифицировать их на отдельные единицы с помощью ДНК-диагностики. Сегодня изучено более 13 генов, мутации которых приводят к развитию доминантных атаксий. Заболевания получили названия согласно порядковому номеру гена, на котором обнаружили аномалии: доминантная спиноцеребеллярная атаксия 1 типа, 2 типа, 3 типа,…, 13 типа и т.д.
Причины
Генная мутация приводит к синтезу нерастворимых внутриклеточных молекул, которые вызывают гибель клетки. При каждом типе доминантной атаксии поражаются определенные нервные клетки.
Симптомы
Вне зависимости от типа атаксии, в клинической картине на первый план выступает поражение мозжечка и его связей с другими отделами ЦНС. Выявляют мозжечковую атаксию, мозжечковую дизартрию и другие симптомы.
Доминантные формы атаксий отличаются друг от друга по дебюту симптомов и течению процесса.
Диагностика
- КТ или МРТ (выявляет специфические изменения в головном мозге, исключает другие заболевания).
- ДНК-тестирование (основной метод диагностики).
Лечение
На сегодняшний день специфическая, эффективная терапия отсутствует.
В качестве вспомогательного лечения применяется ЛФК, вестибулярная гимнастика.
В эту группу входят дентаторубропаллидолюисова атрофия (ДРПЛА), эпизодические (периодические, пароксизмальные) атаксии, синдром Маринеску-Шегрена .
Заболевания встречаются крайне редко, лечение только симптоматическое.
Лекция специалиста на тему атаксия Фридрейха:

Спиноцеребеллярные атаксии – группа генетически разнородных наследственных заболеваний неврологического характера, которые проявляются различными расстройствами работы мозжечка и иногда базальных ядер головного мозга. Симптомами этого состояния являются: развитие атаксии и неустойчивой походки, нарушение координации движений и другие неврологические проявления. Диагностика спиноцеребеллярных атаксий производится на основании данных неврологического осмотра, изучения наследственного анамнеза больного, магнитно-резонансной томографии и молекулярно-генетических исследований. Специфического лечения этой патологии на сегодняшний момент не существует, для сохранения оптимального качества жизни больного используют методы поддерживающей и симптоматической терапии.
- Причины и классификация спиноцеребеллярных атаксий
- Симптомы спиноцеребеллярных атаксий
- Диагностика и лечение спиноцеребеллярных атаксий
- Прогноз и профилактика спиноцеребеллярных атаксий
- Цены на лечение
Общие сведения
Спиноцеребеллярные атаксии – группа наследственных неврологических состояний, характеризующихся развитием прогрессирующей дегенерации клеток мозжечка и иногда базальных ядер вплоть до их полной атрофии. Впервые одно из заболеваний этой группы было описано еще в 1891 году немецким невропатологом П. Менцелем, который выявил развитие атаксии, офтальмоплегии и других неврологических нарушений в рамках одной семьи. Дальнейшие исследования показали, что это состояние (известное сейчас как спиноцеребеллярная атаксия 1-го типа) наследуется по аутосомно-доминантному механизму.
Причины и классификация спиноцеребеллярных атаксий
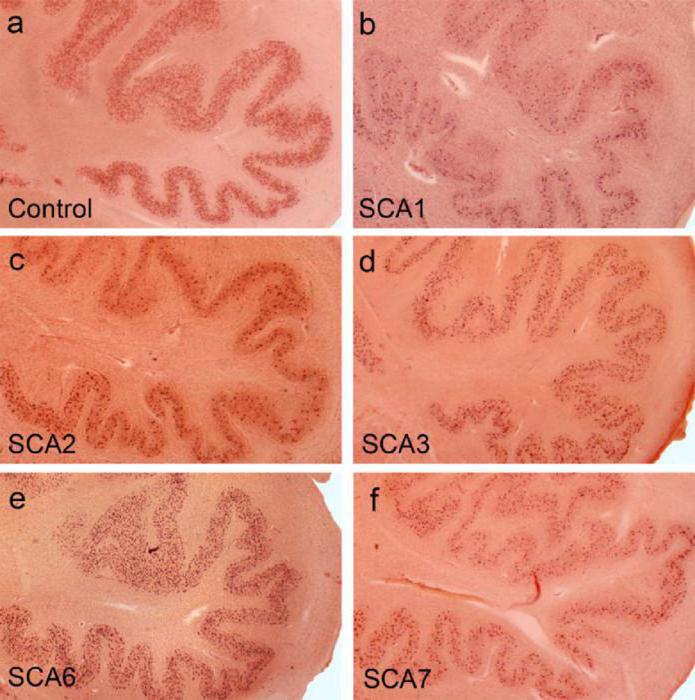
Несмотря на значительное генетическое и отчасти клиническое разнообразие спиноцеребеллярных атаксий, молекулярные механизмы генетических нарушений при этих заболеваниях очень сходны. Основная причина патологии заключается в изменении количества тринуклеотидных последовательностей (CAG) в кодирующей части ассоциированных с заболеванием генов. Это приводит к увеличению количества аминокислоты глутамина в полученном белке, что изменяет физико-химические свойства протеина и нарушает его функции. В ряде случаев вышеуказанные белки прямо или косвенно участвуют в метаболизме нервной ткани, поэтому изменение их структуры приводит к спиноцеребеллярной атаксии. В настоящее время лучше всего изучены молекулярные механизмы 6 основных разновидностей этого заболевания – данные формы патологии встречаются наиболее часто и в совокупности составляют более 90% случаев спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 1-го типа считается самым распространенным и самым изученным вариантом данной патологии. Ее причиной выступают мутации в гене ATXN1, который располагается на 6-й хромосоме. В норме данный ген имеет не более 36 CAG-повторов, увеличение их количества приводит к развитию заболевания. Продуктом экспрессии гена ATXN1 является особый ДНК-связывающий белок, активно участвующий в метаболизме клеток Пуркинье мозжечка – при наличии мутантной разновидности гена это приводит к появлению агрегантов и постепенной дегенерации, что и становится причиной спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 2-го типа – менее распространенный вариант заболевания, этиология не так тщательно изучена. Причиной патологии является увеличение количества CAG-повторов в гене ATXN2, локализованном на 12-й хромосоме. В здоровом варианте гена количество вышеуказанных последовательностей составляет от 15 до 36, тогда как при спиноцеребеллярной атаксии их может быть свыше 100. Функции белка, который кодируется геном ATXN2, на сегодняшний момент неизвестны.
Спиноцеребеллярная атаксия тип 3 (другое название – болезнь Мачадо-Джозефа в честь двух больных, у которых впервые было описано данное состояние) – причиной этого варианта патологии выступают нарушения в гене ATXN3, расположенном на 14-й хромосоме. В норме количество CAG-повторов в этом гене не превышает 47, при развитии заболевания обнаруживается от 53 до 68 повторов. Данный ген кодирует белок, который предположительно участвует в энергетическом обмене нейронов мозжечка и базальных ядер.
Спиноцеребеллярная атаксия тип 6 – сравнительно редкий вид заболевания, обусловленный дефектами в гене CACNA1A, локализованном на 19-й хромосоме. Для развития патологии достаточно очень незначительного увеличения количества CAG-повторов – если в нормальном варианте гена их обнаруживают 5-20, то при наличии атаксии – 21-26. Ген CACNA1A кодирует белок-субъединицу кальциевых каналов, расположенных на нейронах мозжечка. Помимо спиноцеребеллярной атаксии, нарушения в гене CACNA1A обуславливают развитие эпизодической атаксии и некоторые наследственные формы мигрени.
Спиноцеребеллярная атаксия тип 7 – данная разновидность патологии вызывается нарушениями структуры гена ATXN7, который располагается на 3-й хромосоме. У здорового человека количество CAG-повторов составляет не более 35, тогда как при заболевании их количество может достигать нескольких сотен. Функции белка, который кодирует ген ATXN7, на сегодняшний момент изучаются.
Спиноцеребеллярная атаксия тип 8 обусловлена генетическим дефектом гена ATXN8, расположенного на 13-й хромосоме. Как и в других случаях, суть генетического дефекта при этом состоянии заключается в изменении количества тринуклеотидных последовательностей CAG – обычно их около 15-50, тогда как при патологии количество повторов может составлять свыше 1200.
Практически при любом типе спиноцеребеллярной атаксии патологическая форма белка, чрезмерно богатая глутамином, формирует отложения в ядрах или цитоплазме нейронов мозжечка и базальных ядер в виде плотных агрегатов. Этот процесс идет тем быстрее, чем сильнее количество CAG-повторов в ключевом гене отличается от нормы. Этим же объясняется механизм антиципации симптомов спиноцеребеллярной атаксии – в процессе мейоза при образовании половых клеток количество вышеуказанных тринуклеотидных последовательностей может увеличиваться, что приводит к усилению симптомов.
Симптомы спиноцеребеллярных атаксий
Несмотря на значительное генетическое разнообразие спиноцеребеллярных атаксий, проявления разных типов этого заболевания в целом сходны и различаются только второстепенными деталями – возрастом манифестации, особенностями некоторых симптомов. Практически все формы патологии не регистрируются в детском возрасте – лишь отдельные случаи 1 и 2-го типов были замечены у детей младше 7 лет, средний возраст их манифестации – 18-30 лет. Спиноцеребеллярные атаксии 3, 6 и 7-го типов характеризуются еще более поздним развитием – их манифестация практически всегда происходит у лиц старше 30 лет. Нередко подобные нарушения выявляются и у пожилых людей, что затрудняет дифференциальную диагностику этого состояния с болезнью Паркинсона и другими нейродегенеративными заболеваниями старшего возраста.
Чаще всего развитие спиноцеребеллярной атаксии начинается с появления простой неуклюжести в движениях, особенно при ходьбе, беге. В дальнейшем возникает тремор рук, нарушения походки, паралич глазодвигательных мышц (офтальмоплегия), изменяется почерк больного (становится крупнее, строки неровные). В конечном итоге заболевание приводит к выраженной мозжечковой атаксии, расстройствам пирамидальных и экстрапирамидальных путей, паркинсонизму. Некоторые формы патологии характеризуются выраженными нарушениями зрения – развитием атрофии зрительного нерва, пигментной дегенерации сетчатки и других процессов.
Спиноцеребеллярная атаксия 6, 7 и 8-го типов также проявляется расстройствами речи (дизартрия) и глотания, что является причиной затрудненного питания и истощения больных. Именно это обстоятельство и связанные с ними нарушения (например, атрофия мозжечка, сердечная недостаточность) часто становятся причиной смерти пациентов. В зависимости от формы заболевания, объема поддерживающего лечения и качества ухода за больными продолжительность жизни при спиноцеребеллярной атаксии может составлять от 10 до 25 лет с момента возникновения первых симптомов патологии.
Диагностика и лечение спиноцеребеллярных атаксий
Выявление спиноцеребеллярной атаксии производится на основании данных неврологического осмотра, изучения наследственного анамнеза, магнитно-резонансной томографии головного мозга и молекулярно-генетических исследований. При осмотре больных на разных стадиях развития патологии определяются различные по выраженности неврологические нарушения – тремор конечностей, атаксия, изменения речи и голоса, на конечных этапах – дисфагия. Некоторые формы спиноцеребеллярной атаксии сопровождаются достаточно быстрым развитием нарушений зрения, приводящим к полной слепоте. Многолетнее наблюдение за такими больными подтверждает неуклонно прогрессирующее течение заболевания. При изучении наследственного анамнеза могут определяться характерные признаки спиноцеребеллярной атаксии – аутосомно-доминантное наследование, наличие антиципации при передаче болезни от отца.
На МРТ головного мозга при спиноцеребеллярной атаксии обнаруживаются очаги демиелинизации и нейродегенерации в области полушарий, червя мозжечка и базальных ядер. На терминальных стадиях развития заболевания может отмечаться полная атрофия мозжечка. Молекулярно-генетические исследования при спиноцеребеллярной атаксии сводятся к поиску патологически увеличенного количества CAG-повторов в генах, ассоциированных с этим заболеванием. В настоящее время большинство лабораторий мира осуществляет поиск этого дефекта в генах, наиболее часто приводящих к развитию патологии – ATXN1, ATXN2, ATXN3, ATXN7, ATXN8 и CACNA1A.
Специфическое лечение патологии отсутствует, поддерживающая терапия способна несколько замедлить развитие спиноцеребеллярной атаксии, но единого мнения по поводу ее эффективности на сегодняшний момент нет. Применяют витаминотерапию (Е, А, группы В), ноотропные средства, стимуляторы обмена веществ (рибоксин) и метаболизма в нервной ткани. При развитии непроизвольных движений рекомендуют использовать клоназепам и галоперидол. Важную роль в сдерживании прогрессирования спиноцеребеллярной атаксии играет лечебная физкультура – регулярное выполнение правильно подобранного комплекса упражнений позволяет укрепить мышцы и снизить выраженность расстройств равновесия. С этой же целью рекомендуют проведение сеансов лечебного массажа, процедуры электромиостимуляции.
В долгосрочной перспективе прогноз любой формы спиноцеребеллярной атаксии неблагоприятный – это заболевание характеризуется выраженным прогрессирующим течением и со временем приводит сначала к инвалидизации, а затем к смерти больного. Однако в конкретном случае прогноз может быть и менее негативным – например, при развитии патологии в пожилом возрасте и своевременно начатом поддерживающем лечении большинство тяжелых симптомов попросту не успеет проявиться. Если спиноцеребеллярная атаксия возникла в молодом или детском возрасте, продолжительность жизни таких больных даже при интенсивном лечении и тщательном уходе будет резко снижена. Профилактика осуществляется методом медико-генетического консультирования родителей, наследственный анамнез которых отягощен по этому состоянию, и генетической пренатальной диагностики. При этом необходимо учитывать аутосомно-доминантный характер наследования спиноцеребеллярной атаксии и такие особенности ее передачи, как антиципация.
Редкой формой наследственных болезней является спиноцеребеллярная атаксия (СЦА). Она проявляется нарушением координации. Кроме того, клинику дополняют другие симптомы неврологии. Лечения патологии не существует.
Этиология
Причиной болезни становится мутирующий ген. Он передается аутосомно-доминантно. Важно, что всего одна копия родительского гена вызывает заболевание. Диагноз ставят на основе изучения генотипа. Спиноцеребеллярная дегенерация имеет около 20 форм.
Точный механизм гибели Нейрон
Узко специализированная клетка, которая является структурной единицей нервной системы.
Белок, участвующий в процессе деградации других белков. При присоединении убиквитина к молекуле происходит ее уничтожение ферментами протеазами.
Узко специализированная клетка, которая является структурной единицей нервной системы.
Заболевания, связанные с прогрессивной гибелью нейронов. Например, болезнь Паркинсона, Альцгеймера, хорея Гентингтона.

Это десятый международный пересмотр статистической классификации болезней и проблем, связанных со здоровьем.
Причины
Выделить причинный фактор ученым не удалось.
Возможно, на развитие атаксии влияют:
Алкоголь высокотоксичен. Большие дозы спирта разрушают клетки Мозжечок
Отдел мозга, отвечающий за равновесие, координацию, мышечный тонус.
Дополнение! Известно, что атаксия – это потеря контроля за равновесием и нарушение координации движений.
Гормоны щитовидной железы влияют на синтез миелина. Известно, что гипотиреоз препятствует восстановлению нейронов. Кроме того, они влияют на деление клеток мозжечка. Как правило, опухоли сдавливают ткань в результате роста. В Мозжечок
Отдел мозга, отвечающий за равновесие, координацию, мышечный тонус.

Вестибулярная атаксия
Классификация
Принято различать врожденные и приобретенные поражения мозжечка. Врожденные атаксии классифицируют согласно типу наследования.
Сидеробластная анемия с СЦА
Рецессивный сцепленный с полом (ХР)
Атаксический синдром появляется в результате поражения:
- Задних столбов спинного мозга (сенситивный);
- Мозжечок
Отдел мозга, отвечающий за равновесие, координацию, мышечный тонус.
Cимптомы
Для всех типов болезни характерны следующие признаки:
Это нарушенная нечеткая речь.
Усиление работы оральных мышц.
Скандирование фраз. Смена звука.
Больной не может определить расстояние до предмета.
Непроизвольное дрожание любой из конечностей или головы, тела, туловища.
Возникает и в покое, и в движении.
Как правило, этим отличается от болезни Паркинсона.
Непроизвольные движения глазных яблок с высокой частотой. Выделяют физиологический и патологический нистагм.
Глаза хаотично двигаются.
Моторика не проходит и в покое, когда человек спит.
Нельзя четко смотреть на конкретный предмет.
Изменения связанные с нарушением удержания позы. Постуральная неустойчивость характеризуется шаткостью при ходьбе, падениями, неспособностью удерживать равновесие.
Пациент не может удержать позу.
Часто случаются падения.
Первый вид. Спиноцеребеллярная атаксия 1 типа протекает тяжело. Как правило, симптомы включают Тремор
Непроизвольное дрожание любой из конечностей или головы, тела, туловища.
Второй вид. СЦА 2 типа характерна для молодых людей. Однако, возможны и поздние случаи развития недуга. Кроме атаксии у пациентов, появляются аномальные движения глаз. Их называют саккады. Мутация расположена в гене ATXN2 на 12 хромосоме. Патология не имеет лечения.
Пятый и шестой вид. СЦА 6 передается аутосомно-доминантным путем. Мутантный ген CACNA 1A на 19 хромосоме. Во время болезни преобладают нарушения походки. Пациент через несколько лет теряет способность ходить. Другие признаки появляются редко. Для СЦА 5 появляется дефект ферментных систем клетки.
Седьмой вид. Болезнь проявляется атаксией и утратой зрения. Известно, что активные симптомы появляются в зрелом возрасте (старше 30 лет). Идет активная гибель клеток желтого пятна. Мутированный ген ATXN 7 лежит в 3-ей хромосоме.
Наследственные спиноцеребеллярные атаксии
Наследственная атаксия, обусловленная нехваткой витамина Е
Это редкая форма наследственных болезней. Появляется она в раннем возрасте (от 4-18 лет). Как правило, этот вид атаксий выявляют в Северной Африке и странах Средиземноморья. Симптомы включают поражение мимики и речи. Позже подключаются изменения скелета и сердца. Аналогично протекает атаксия Фридрейха. Дефект гена расположен в длинном плече 8 хромосомы.
Аутосомно-доминантные спиноцеребеллярные атаксии
К этой группе относятся 12 форм заболевания (СЦА 1,2,3,6,7,8,10,12,17, 31,36 и ДРПЛА). Такой тип наследования дает 50% передачу недуга от родителей к детям. Конечно, при условии, что дефектный ген есть у обоих. Иначе, только один из предков способен передать болезнь в 25% случаев. Кроме атаксии, доминируют признаки нарушения зрения, аномальные движений глаз. А также появляются саккады и тремор.
Заболевание появляется в раннем возрасте. Кроме неврологии, в процесс вовлечены другие органы. Так, искривляется позвоночник. Иногда деформируется стопа. В редких случаях недуг захватывает мышцу сердца. От патологии Фридрейха страдают гормоны. Часто поражаются половые железы. В конце болезни пациент выходит на сахарный диабет.
Другие формы наследственных спиноцеребеллярных атаксий
Разновидностей атаксий довольно много (болезнь Вереллея, атаксия Пьера-Мари, синдром Фуа-Алажуанина). И различать формы помогают следующие признаки:
- Возраст дебюта;
- Характер проявлений;
- Течение болезни.
К примеру, атаксия Пьера-Мари напоминает болезнь Фридрейха. Она была выделена в отдельную форму в начале прошлого века. Недуг начинается поздно. Как правило, первые признаки заметны в 30-40 лет. Например, при патологии нет костных деформаций. Доминируют признаки Дисфагия
Нарушение нормального глотания.
Синдром Мари-Фуа-Алажуанина встречается крайне редко. Известно, что начало у болезни позднее (средний возраст 47 лет). Как правило, гибнут клетки червя мозжечка и волокон Пуркинье. Характерно медленное течение. Таким образом, полный паралич наступает только после 15-20 лет от начала симптомов.
Диагностика
Постановка диагноза при атаксии опирается на:
- Клинический осмотр;
- Генеалогию;
- Результаты генетической диагностики;
- Визуализацию.
Метод диагностики, при котором исключаются заболевания, не подходящие по определенным критериям.
Болезнь носит семейный характер. Типы наследования разнообразны. Это сцепленный с полом, рецессивный и доминантный вид. Установить форму наследования поможет генеалогический анализ.
Генетический тест наиболее информативен. Он поможет выявить мутированные гены. Установить количество повторов ЦАГ в ATXN. Как правило, для анализа берут соскоб с внутренней поверхности щеки. Но можно использовать любой био-материал.

Диагностика спиноцеребеллярной атаксии
Метод обследования внутренних органов и тканей с помощью явления ядерного магнитного резонанса.
Методика МРТ, с помощью которой можно увидеть проводящие пути головного мозга (тракты).
Неинвазивный метод исследования головного мозга, при котором регистрируется его биоэлектрическая активность.
Лечение
Основные задачи терапии:
Непроизвольное дрожание любой из конечностей или головы, тела, туловища.
Лечение для спиноцеребеллярной атаксии не существует. Якутские ученые тестируют L-цистеин в качестве терапии СЦА 1. К сожалению, достоверных данных о его эффективности пока нет. В 2017 году завершена 2 фаза испытания препарата Trehalose для СЦА 3 типа. Он одобрен FDA. А также он имеет статус орфанного лекарства. Кроме того, продолжаются исследования в области генной инженерии.
N-ацетил-L-лейцин – это модифицированная аминокислота. Ее синтезировали ученые из Британии. В 2017 году лейцин стали применять для лечения СЦА. У препарата хороший профиль безопасности. Лечение легко переносится пациентами.
Для пациента с атаксией важно сохранить активность. Занятия с инструктором ЛФК позволяют тренировать походку. Врач дает советы, как удерживать позу, выходить на ежедневную прогулку. Эрготерапевт поможет организовать интерьер дома. Регулярные занятия укрепляют мышцы. Упражнения полностью компенсируют дефицит моторики при легких степенях атаксии.
Прогноз излечения, продолжительности жизни, профилактика, инвалидность
На сегодняшний день нет данных об успешных случаях терапии СЦА. Известно, что несколько лекарств проходят испытания. Результаты тестов будут опубликованы в ближайшие пять лет. К сожалению, пока остается только симптоматическое лечение.

Лечение спиноцеребеллярной атаксии
Пациенты с СЦА живут 8-14 лет с момента начала заболевания. Обычно смерть наступает в 58-62 года. На продолжительность жизни влияют:
- Число повторов ЦАГ;
- Сопутствующие заболевания;
- Условия жизни;
- Активная реабилитация.
Специфической профилактики болезни нет. При выявлении атаксии в семье обследуются близкие родственники. Как правило, это родители, братья и сестры. Известно, планирование беременности в таких семьях начинается с наследственного анализа. Во время вынашивания проводится пренатальная диагностика.
Инвалидность дает возможность социальной поддержки. Группа позволяет больному бесплатно или со льготами получать средства технической реабилитации, лекарства. При невозможности самостоятельного обслуживания МСЭ устанавливает первую группу. При легких стадиях атаксии возможно получение 2 или 3-ей группы.
Читайте также:
