
Абсанс эпилепсия или миоклоническая эпилепсия
Carlo Alberto Tassinari, Guido Rubboli, Roberto Michelucci
История и номенклатура
Первые сообщения об абсансных приступах, которые сопровождаются значительными клоническими и миоклоническими сокращениями, появились в 1966 году (Gibberd 1966). Только спустя несколько лет миоклонические абсансы стали рассматриваться как специфический тип приступов (Tassinari et al 1969), а позже было предложено выделить их в качестве отдельного синдрома (Tassinari and Bureau 1985).
Средний возраст начала эпилепсии с миоклоническими абсансами составляет 7 лет (диапазон от 2 до 12.5 лет). Вначале указывалось на преобладание пациентов мужского пола, однако в другом исследовании наблюдалось преобладание девочек (Manonmani and Wallace 1994). Примерно у половины детей с такими приступами отмечается задержка психического развития, еще до времени развития приступов (Tassinari and Bureau 1985; Tassinari et al 1992).
Заболевание проявляется внезапными абсансами, которые сопровождаются билатеральными выраженными миоклониями. Нарушение сознания может быть полным или частичным. Приступы вовлекают в основном мышцы плечевого пояса, рук и ног, реже – лица. Если возникают лицевые миоклонии, они наиболее выражены вокруг рта и подбородка, при том, что моргания век как правило отсутствуют или крайне редки. Двигательные расстройства могут носить продолженный и прогрессирующий характер, могут сочетаться с тоническим сокращением, которое максимально в плечевом поясе, дельтовидной мышце. Миоклонии и тонические сокращения могут быть симметричными или доминировать с одной стороны, что может приводить к повороту головы и туловища. Могут присутствовать вегетативные проявления, такие как остановка дыхания, недержание мочи (Tassinari and Bureau 1985; Tassinari et al 1992).
Каждый эпизод миоклонического абсанса длится от 10 до 60 секунд. Приступы могут возникать неоднократно в течение дня. Гипервентиляция, пробуждение и ритмическая фотостимуляция (РФС) могут провоцировать атаки. Однако во время сна миоклонические приступы урежаются по мере нарастания глубины сна (Tassinari and Bureau 1985; Tassinari et al 1992). Описаны редкие случаи эпилептического статуса миоклонических абсансов (Manonmani and Wallace 1994; Tassinari et al 1995).
Примерно в 2/3 случаев отмечаются другие виды приступов – генерализованные тонико-клонические (ГТКП), чистые абсансы, приступы падений (Tassinari and Bureau 1985; Tassinari et al 1992).
Этиология эпилепсии с миоклоническими абсансами неизвестна. Примерно в четверти случаев в семейном анамнезе отмечаются эпилептические приступы, однако генетические факторы и наследственные механизмы неизвестны. В одном из последних исследований у 7 из 14 пациентов с миоклонической абсанс эпилепсией обнаружены хромосомные нарушения (трисомия 12 p и синдром Ангельмана) (Elia et al 1998a; 1998b).
Патогенез и патофизиология
В настоящее время четкие данные по патогенезу эпилепсии с миоклоническими абсансами отсутствуют (Tassinari and Bureau 1985; Tassinari et al 1992). Последние исследования, указывающие на хромосомной дисфункцию, могут свидетельствовать о возможной роли в патогенезе аномальной экспрессии генов пораженной хромосомы (Elia et al 1998a; 1998b).
Синдром встречается редко. Точные данные по частоте заболевания неизвестны (Tassinari and Bureau 1985; Tassinari et al 1992), имеются результаты наблюдений из центра St. Paul в Марселе, которые дают цифры 0.5%-1% среди всех больных эпилепсией, обратившихся в этот центр (Tassinari and Bureau 1985).
Абсансы при этом синдроме похожи на абсансы при детской (ДАЭ) или юношеской (ЮАЭ) абсанс эпилепсиях. Несмотря на то, что при абсансы при ДАЭ и ЮАЭ могут также включать миоклонические компоненты, они не генерализованные, вовлекают в основном веки или лицевую мускулатуру, гораздо менее интенсивные, чем эпилепсии с миоклоническими абсансами (Tassinari and Bureau 1985; Tassinari et al 1992). Помимо этого, при ней гораздо чаще, чем при ЮАЭ, встречается задержка психического развития.
Периоральный миоклонус с абсансами, описанный Panayiotopoulos (Panayiotopoulos et al 1994), имеет черты, схожие с эпилепсией с миоклоническими абсансами, в том числе эволюционные характеристики, поэтому может рассматриваться как вариант этого синдрома (Tassinari et al 1996). Дифференциальный диагноз должен включать также генерализованные миоклонии, для которых нехарактерно нарушением сознания и которые сопровождаются отчетливыми разрядами полиспайк-волн. Частые асимметричные моторные проявления при миоклонических абсансах могут напоминать картину парциальных моторных приступов. В таких случаях постановке точного диагноза может помочь ЭЭГ и полиграфическая регистрация приступов.
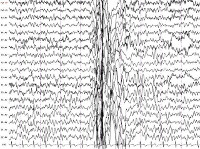
Для иктальной ЭЭГ при эпилепсии с миоклоническими абсансами характерен паттерн билатерально-синхронных и симметричных разрядов комплексов спайк-волна частотой 3 Гц, который напоминает похожий паттерн при детских абсансах (Tassinari and Bureau 1985; Tassinari et al 1992). Завершение разрядов может сопровождаться дельта волнами в лобных отделах, нередко асимметричными. Комплексы спайк-волна могут перемежаться с полиспайк-волновыми комплексами (Tassinari et al 1995). Полиграфическая регистрация миоклонических абсансов показывает, что билатеральные миоклонии имеют ту же частоту, что и разряды спайк-волн, начинаются с задержкой 1-2 секунды, после этого развивается тоническое, иногда асимметричное сокращение, максимальное в плечевом поясе (Tassinari and Bureau 1985; Tassinari et al 1992). Показана также строгая корреляция позитивного компонента спайка и миоклонии (Tassinari et al 1969). Интериктальная ЭЭГ в основном характеризуется нормальной картиной основной активности, на которую наложены генерализованные спайки и волны, реже – фокальные или мультифокальные спайки и волны. Фотосенситивность нехарактерна. ЭЭГ сна демонстрирует нормальную организацию и симметричные физиологические паттерны. Эволюция спайк и волн при смене стадий сна в целом походит на эволюцию спайков и волн, наблюдаемую при детской абсанс эпилепсии (Tassinari and Bureau 1985; Tassinari et al 1992;1996).
Прогноз и осложнения
Длительное течение миоклонических абсансов вероятно играет основную роль в появлении психической задержки – поскольку психические функции у детей с быстрой ремиссией не нарушаются. Ряд авторов считает, что задержка психического развития может быть вызвана непосредственно текущей приступной активностью (Manonmani and Wallace 1994).
В редких случаях прекращение миоклонических абсансов сопровождается развитием других типов приступов, таких как атипичные абсансы или субклинические тонические приступы, что может сформировать клиническую картину, напоминающую синдром Леннокса-Гасто (Tassinari and Bureau 1985; Tassinari et al 1992).
Комбинированное применение вальпроатов и этосуксимида в высоких дозах (при контроля уровня в плазме от 80 до 130 мкг/мл, от 70 до 110 мкг/мл соответственно) является методом выбора при эпилепсии с миоклоническими абсансами. В отдельных случаях хорошие результаты могут быть достигнуты политерапией, включающей фенобарбитал, вальпроаты и бензодиазепины (Tassinari et al 1992). Ряд авторов отмечают положительный эффект монотерапии ламотриджином, если же он неэффективен – назначения вальпроатов как второго препарата (Appleton 1994; Manonmani and Wallace 1994).
Список литературы
Appleton RE. Epilepsy with myoclonic absence. Arch Dis Child 1994;71:180.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
Elia M, Guerrini R, Musumeci SA, Bonanni P, Gambardella A, Aguglia U. Myoclonic absence-like seizures and chromosome abnormality syndromes. Epilepsia 1998a;39:660-3.
Elia M, Musumeci SA, Ferri R, Cammarata M. Trisomy 12p and epilepsy with myoclonic absences. Brain Dev 1998b;20:127-30.
Gibberd FB. The clinical features of petit mal. Acta Neurol Scand 1966;42:176-90.
Manonmani V, Wallace SJ. Epilepsy with myoclonic absences. Arch Dis Child 1994;70:288-90.
Panayiotopoulos CP, Ferrie CD, Giannakodimos S, Robinson RO. Perioral myoclonia with absences: a new syndrome? In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey, 1994:143-53.
Tassinari CA, Bureau M. Epilepsy with myoclonic absences. In: Roger J, Dravet C, Bureau M, Dreifuss FE, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey, 1985:121-9.
Tassinari CA, Bureau M, Thomas P. Epilepsy with myoclonic absences. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey, 1992:151-60.
Tassinari CA, Lyagoubi S, Santos V, et al. Etude des decharges de pointes ondes chez l'homme. II. Les aspects cliniques et electroencephalographiques des absences myocloniques. Rev Neurol 1969;121:379-83.
Tassinari CA, Michelucci R. Epilepsy with myoclonic absences: a reappraisal. In: Wolf P, editor. Epilepsic seizures and syndromes. London: John Libbey, 1994:137-41.
Tassinari CA, Michelucci R, Rubboli G, et al. Myoclonic absence epilepsy. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. Guildford: Churchill Communications Europe, 1995:187-95.
Tassinari CA, Rubboli G, Michelucci R. Epilepsy with myoclonic absences. In: Wallace J, editor. Epilepsy in children. London: Chapman & Hall, 1996:287-91.
Данный недуг является одним из самых распространенных неврологических заболеваний. В той или иной мере симптомы расстройства проявляются примерно у 10% людей. У детей недуг встречается в несколько раз чаще, чем у взрослых, причем протекает лечение эпилепсии абсансной значительно сложнее.

Что такое абсансная эпилепсия
Абсансная эпилепсия является генерализованным припадком, встречающимся чаще всего у детей, возрастом от 3-х о 14-ти лет. Со стороны его выявить непросто, так как он больше похож на задумчивость и мечтательность.
Что такое абсансы при эпилепсии у взрослых и детей мало кто знает. Характеризуются они непродолжительным по времени помутнением рассудка, сопровождающимся остановившимся взглядом в одну точку, частым морганием или закатыванием зрачков кверху. Пациент может испытывать от 50 до 100 эпизодов в сутки. У многих юных пациентов данный тип заболевания проходит самостоятельно к наступлению переходного возраста.
Причины возникновения абсансной эпилепсии у взрослых
Вопрос этиологии заболевания, а в особенности абсансов, далек от решения. Исследователями прослеживаются генетические механизмы возникновения болезни. В качестве пускового механизма бессудорожной невралгии абсансной эпилепсии у взрослых большое значение играют:
- травмы черепа;
- опухоли мозга и другие тяжелые заболевания;
- злоупотребление спиртными напитками, наркотическими веществами, табакокурением;
- наследственный фактор.
Симптомы абсансной эпилепсии
Главная сложность, связанная с бессудорожной абсансной эпилепсией, — риск спутать болезнь с задумчивостью, из-за чего недуг зачастую остается без должного внимания. Симптомы абсансной эпилепсии у взрослых не всегда ясны, но основные признаки включают:
- расфокусированность взгляда;
- отсутствие отклика на внешние воздействия;
- остановка предложения на полуслове в момент приступа;
- спутанные движения конечностями;
- моргание;
- бесцельное брождение.
Какие абсансы бывают
Характерной чертой является высокая периодичность, в особо напряженных ситуациях достигающая нескольких десятков и сотен припадков в сутки. Провоцируют их следующие явления:
- активная умственная деятельность;
- чрезмерное расслабление;
- гипервентиляция легких;
- недосып;
- вспышки света;
- мерцающий экран телевизора или компьютера.
Абсансы проявляются следующей клинической картиной простых эпизодов:
- длятся считанные секунды;
- у пострадавшего отсутствует реакция, и он находится в бессознательном состоянии;
- человек не замечает приступа.
Пароксизм может длиться 5-30 секунд, в течение которых пациент теряет осознанное восприятие окружающего мира. Со стороны заметно отсутствие осознанного взгляда, выключение человека из деятельности и непродолжительное застывание на месте. Выраженный эпизод обусловлен остановкой начатой речи или действий, а также заторможенным продолжением, идущим перед приступом активности.
В первом случае после возникновения пароксизма наблюдается восстановление двигательной активности и слов ровно с того эпизода, где они остановились. Пострадавшие характеризуют данное состояние, как резкий ступор, провал в памяти, выпадение из реальности, транс. В послеприступное время самочувствие у больного нормальное.
Другой вид припадка является более заметным и для пострадавшего, и для окружающих, так как выделяется двигательными и тоническими феноменами. Пароксизм протекает с уменьшением мышечной активности, что приводит к пониканию головы и ослаблению конечностей. В редких случаях больной может сползать со стула, а при тотальной атонии падать.
Тонические пароксизмы сопровождаются гипертонусом мышц. В зависимости от локализации очагов может быть заметно:
- изгибание тела;
- сгибание и разгибание конечностей;
- запрокидывание головы.
Приступ с миоклоническим компонентом характеризуется низкоамплитудными сокращениями мышц в виде частых подергиваний телом. У взрослого человека может возникать подергивание уголков рта, подбородка, век. Миоклонии бывают:
- симметричными;
- ассиметричными.
Возникающие во время эпизода автоматизмы могут нести характер элементарных, но повторяющихся движений:
- бормотание;
- сглатывание;
- пережевывание;
- потирание рук;
- покачивание ногой;
- застегивание кнопок или пуговиц.
Частота сложных может варьироваться от нескольких до десятков раз в день. Эпизоды могут быть единственным проявлением у пациента, что больше характерно для детей.
Бессудорожная абсансная эпилепсия
Патология гораздо чаще возникает в раннем возрасте, от 7-ми до 14-ти лет, и в молодости — от 15-ти до 30-ти лет. До четырехгодовалого возраста у пациентов простые абсансы не возникают, так как для проявления данного феномена необходима определенная зрелость головного мозга.
Пусковым механизмом бессудорожной невралгии являются:
- нейроинфекция;
- травмы черепа.
Во время бессудорожных пароксизмов могут возникать вкусовые, обонятельные и зрительные галлюцинации. Характерной особенностью болезни считается большое количество клинических проявлений. Поэтому органическое заболевание различной этиологии, при отсутствии квалифицированной помощи, склонно к принятию хронического характера.
Сложные формы абсансной эпилепсии
Сложные формы являются такими состояниями, при которых отмечаются характерные и повторяющиеся для пациента действия или проявления на фоне полной потери сознания. Например, это могут быть автоматические действия, характеризующиеся стереотипными чередующимися или аналогичными движениями:
- движениями глаз, губ или языка;
- жестами;
- привычными действиями, доведенными до автоматизма, расчесыванием, складыванием одежды или канцелярских принадлежностей.
Поэтому пароксизмы сложно отличить от обыденного поведения человека. Также сложные абсансы могут сопровождаться увеличением мышечного тонуса. В таком случае отмечается вытягивание туловища назад, закатывание зрачков, откидывание головы. В более выраженных ситуациях больной может выгибать туловище сзади и делать шаг назад, для сохранения равновесия. Часто помутнение сознания происходит на фоне спада тонуса мышц с последующим падением.
При сложных миоклонических абсансах отмечаются двусторонние ритмические феномены миоклонического характера, зачастую мимической мускулатуры и мышц верхних конечностей. Сложные припадки требуют меньшей зрелости головного мозга, поэтому могут возникать в возрасте 4-5 лет.
Клиническая картина сложных приступов:
- продолжительность более десяти секунд;
- пострадавшего без сознания можно перемещать, при этом передвигается он самостоятельно;
- человек, перенесший пароксизм, понимает, что с ним что-то происходит, дополнительно от отмечает факт помутнения сознания.
Диагностика абсансной эпилепсии
Патология способна проявлять себя в виде совокупности различных признаков. Приступы могут протекать с потерей сознания или без нее. Во время локализации судорожного припадка подергиваются мышцы, ощущаются незнакомые ощущения в теле, неосознанные наплывы мыслей. Дополнительно для каждого возраста характерны определенные особенности возникновения и протекания патологии.
Диагностирование заболевания подразумевает под собой ряд процедур, обычно включающих в себя:
- полный анализ крови;
- электроэнцефалографию;
- компьютерную томографию;
- магнитную резонансную томографию.
Вышеперечисленные методики позволяют доктору выявить причину возникновения недуга, а также определить его вид.
Одним из самых важных этапов диагностики является первичный осмотр у врача. Начинается он с анализа жалоб пострадавшего, как правило основными будут:
- помутнение сознания;
- спазмы мышечной ткани;
- застывание на месте.
Задавая уточняющие вопросы, доктор может выяснить периодичность припадков и то, как они проявляются в конкретном случае.
Для точной постановки диагноза необходимо понимание того, в каких условиях возник пароксизм, какой симптоматикой сопровождался и что происходило после него. Эта информация необходима для проведения дифференциальной диагностики, то есть процесса разграничения схожих патологий и отсеивания неверных вариантов. Потому что присутствие головных болей, искажений восприятия, двигательных нарушений может указывать не только на болезнь, но и на мигрень.
После устного опроса доктор переходит к сбору анамнеза, включающего в себя:
- информацию о случаях патологии у ближних и дальних родственников;
- возрасте начала припадков;
- и о наличии травм головы или сопутствующих недугов.
Уже на этом этапе полученные данные дают возможность неврологу предполагать наличие или отсутствие болезни, а также наметить направление процесса диагностики, предпочтительной терапии. Однако решения должны быть также подкреплены инструментальными и лабораторными исследованиями. Это поможет избежать непреднамеренной ошибки при постановке диагноза.
Анализ крови является одним из самых доступных методов исследования при многих заболеваниях, так как он помогает получить максимально точные данные о состоянии организма человека. Своевременное выявление различных отклонений от нормальных показателей позволяет как можно скорее начать эффективную терапию. Обнаружить заболевание можно путем измерения содержания количества электролитов в плазме крови. Также проводят анализ во время приема противоэпилептических препаратов, чтобы выявить, достигнута ли необходимая концентрация действующего вещества.
Электроэнцефалограмма — это безвредный способ диагностики, необходимый для оценки электрической активности головного мозга. Длительность процедуры составляет от 60 до 90 минут. В процессе исследования на голову пациента помещают специальные электроды, напоминающие металлические круги.
Дополнительно используется методика, когда электроэнцефалограмму проводят во время сна. Это помогает максимально подробно изучить состояние человека. Исследование проводится не только на этапе диагностики, но и в процессе лечения, для контроля эффективности терапии. При наличии диагноза процедура может производиться чаще.
Для обнаружения структурных нарушений головного мозга используются методы нейровизуализации:
- компьютерную томографию;
- магнитную резонансную томограмму.
Данные диагностические процедуры абсолютно безболезненные. Самой дискомфортной частью процесса может стать укол контрастирующего препарата, необходимый для того, чтобы определенные участки тканей были видны на снимке максимально четко. Во время сканирования пациенту рекомендуется расслабиться и не совершать никаких движений.
Лечение абсансной эпилепсии
Основной упор при лечении патологических факторов делается на медикаментозную терапию препаратами, относящимися к противосудорожной и противоэпилептической группе. Подбор лекарств должен производиться непосредственно лечащим врачом, опирающимся на возраст пострадавшего и частоту приступов.
В том случае, если первое средство потеряло эффективность, его необходимо в срочном порядке заменить на другое. Прием одновременно нескольких медикаментов допускается только в той ситуации, если их действие дополняет друг друга.
Прогноз абсансной эпилепсии
При условии применения адекватной терапии лечение болезни проходит успешно. Однозначно ответить на вопрос, излечима ли абсансная эпилепсия, невозможно. Так как зачастую, по мере взросления, недуг трансформируется в устойчивую ремиссию, и не напоминает о себе. При миоклонических судорогах высокой частотности, субнормальном интеллекте и резистентности к терапии медикаментами прогноз хуже.
Отмена лекарственных средств осуществляется поэтапно, только после консультации с неврологом и при продолжительном периоде отсутствия приступов.

Миоклоническая эпилепсия — заболевание, основу которого оставляют миоклонические эпилептические пароксизмы. Эпизоды миоклонических судорог у больных сочетаются с генерализованными клонико-тоническими эпиприступами, абсансами. Сопутствующая неврологическая симптоматика зависит от формы эпилепсии. Диагностика включает сбор анамнеза, оценку неврологического и психического статуса, электроэнцефалографию, генеалогический анализ, биохимические исследования, нейровизуализацию. Лечение проводится антиконвульсантами, при резистентности — комбинацией противоэпилептических препаратов.
- Причины миоклонической эпилепсии
- Патогенез
- Классификация
- Симптомы миоклонической эпилепсии
- Осложнения
- Диагностика
- Лечение миоклонической эпилепсии
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Причины миоклонической эпилепсии
Обычно ведущим является генетический фактор. Чёткое аутосомно-доминантное наследование прослеживается при синдроме Драве, аутосомно-рецессивное — в отдельных случаях ранней миоклонической энцефалопатии. Некоторые заболевания имеют полигенное наследование. Локализация генетических дефектов установлена не для всех наследственных форм, исследования в этом направлении продолжаются. К генетически детерминированным патологиям относится и симптоматическая МЭ, возникающая вследствие дисметаболических процессов, обусловленных наличием дефектных генов. Образованию спонтанных мутаций в геноме способствуют:
- Внутриутробные инфекции. Инфекционный процесс неблагоприятно отражается на развитии плода. Особенно опасны вирусные инфекции, поскольку вирусы способны провоцировать аномальную перестройку отдельных генов.
- Хронические заболевания беременной. Сахарный диабет, сердечная недостаточность, хронические заболевания лёгких, эндокринная патология матери приводят к гипоксии, метаболическим расстройствам на ранних стадиях развития зародыша. В результате происходят сбои формирования ЦНС, отдельных механизмов обмена веществ.
- Повышенный радиоактивный фон. Радиация оказывает мутагенное влияние на живые организмы. Развивающийся плод наиболее подвержен подобному воздействию. Следствием является возникновение структурных, дисметаболических, функциональных аномалий, влекущих за собой повышенную эпилептическую активность.
- Прием тератогенных медикаментов. Самолечение, незнание о своей беременности в раннем периоде, медицинская необходимость фармакотерапии приводят к приёму опасных для плода медикаментов. Химические вещества оказывают повреждающее воздействие на отдельные гены, вносят изменения в существующие метаболические механизмы.
- Токсические воздействия на плод.Алкоголизм, наркомания, курение женщины в период беременности сопровождаются проникновением токсических веществ в организм плода. Подобно тератогенным фармпрепаратам они способны повредить отдельный локус генома, в результате возникает миоклоническая эпилепсия.
Патогенез
Идиопатические варианты МЭ развиваются вследствие генетически обусловленной повышенной возбудимости церебральных нейронов, приводящей к эпилептогенной активности мозга. Симптоматическая миоклоническая эпилепсия формируется в результате обменных нарушений, накопления в нервных клетках патологических соединений (полисахаридных включений, прионных белков).
При болезни Лафоры, миоклонической энцефалопатии младенцев повышенная эпиактивность обусловлена дисметаболизмом нейронов в условиях разрастания глиальных элементов (при гибели нейронов, нарушении апоптоза астроцитов). Нейрональная гипервозбудимость вызывает возникновение патологической нервной импульсации, идущей к мышечным волокнам. Результатом являются отдельные мышечные сокращения (миоклонии), тонические, клонические судороги. Различная локализация миоклоний отражает локальное возбуждение разных зон мозговой коры. При диффузном распространении гипервозбуждения возникает клонико-тонический пароксизм с тотальным вовлечением мышечных групп.
Классификация
В основе группировки отдельных видов МЭ лежит этиологический принцип. Согласно Международной классификации эпилепсии 1989 года выделяют 3 основные группы:
- Идиопатические — наследственно обусловленные формы. Характерна манифестация симптоматики в детском/подростковом возрасте. Идиопатическими являются доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ), юношеская миоклоническая эпилепсия (ЮМЭ), болезнь Унферрихта-Лундборга, синдром Драве.
- Криптогенные — не имеющие установленной этиологии. Отличаются выраженной резистентностью к фармакотерапии, наличием сопутствующей очаговой симптоматики, интеллектуального дефицита. К криптогенным относятся эпилепсия с миоклонически-астатическими приступами, эпилепсия с миоклоническими абсансами.
- Симптоматические — возникающие на фоне происходящих в организме патологических процессов. В большинстве случаев обусловлены метаболическими нарушениями. Симптоматическими считаются ранняя миоклоническая энцефалопатия, болезнь Лафоры, миоклонические пароксизмы при подостром склерозирующем панэнцефалите, болезни Крейтцфельдта-Якоба.
Впоследствии были выявлены генетические аспекты возникновения криптогенных форм МЭ. Учитывая результаты исследований, Международное общество неврологов предложило относить ранее считавшиеся криптогенными виды МЭ к идиопатическим.
Симптомы миоклонической эпилепсии
Базовым симптомом выступают пароксизмы миоклоний, затрагивающие различные мышечные группы конечностей, реже — лица, еще реже — туловища. Миоклонии выглядят как мышечные подёргивания, при вовлечении мышц одной группы сокращения приводят к непроизвольным двигательным актам, напоминающим гиперкинезы. Миоклонический эпилептический пароксизм происходит при сохранённом сознании, может протекать с перемещением сокращений по различным мышцам. Миоклоническая эпилепсия характеризуется комбинацией миоклоний с клонико-тоническими приступами и/или абсансами. В зависимости от нозологической формы наблюдаются задержка психического развития, атаксия, пирамидная недостаточность, расстройства мышечного тонуса, зрительные нарушения.
ДМЭМ дебютирует в возрастном периоде от 6 месяцев до 3 лет. Приступы захватывают верхние конечности, лицо, шею, могут имитировать наклон головы, моргание, кивки головой. Заболевание редко сопровождается интеллектуальным снижением. Миоклоническая эпилепсия юношеского возраста (манифестация в возрасте 12-18 лет) отличается присоединением тонико-клонических эпизодов, отсутствием неврологического дефицита. Синдром Драве клинически проявляется на первом году жизни, сопровождается олигофренией, нарушениями поведения, пирамидным дефицитом. Семейная миоклония Унферрихта-Лундборга начинается в 5-16 лет, сочетается с тремором, атаксией, дизартрией, психическими расстройствами.
Миоклоническая эпилепсия симптоматического характера отличается прогрессированием симптоматики, выраженным когнитивным дефицитом, прочими неврологическими нарушениями, наличием проявлений основного заболевания, клинико-лабораторных признаков метаболических расстройств.
Осложнения
Клонико-тонические, астатические приступы осложняются травмированием пациента вследствие падения. Генерализованные судороги с утратой сознания опасны западением языка, перекрытием дыхательных путей и асфиксией. Аспирация слюны, рвотных масс приводит к последующему развитию пневмонии. Длительный миоклонический пароксизм, непрерывно следующие кластерные сокращения перерастают в миоклонический эпистатус. В эпилептическом статусе возможны серьёзные дыхательные расстройства, остановка сердца, развитие отёка головного мозга.
Диагностика
- Сбор анамнестических данных. Большое значение имеет возраст дебюта, характер начала, порядок развития симптоматики.
- Неврологический осмотр. Проводится неврологом, направлен на выявление миоклонических сокращений, очагового дефицита, определение уровня психического развития, степени когнитивных расстройств, оценку психического статуса.
- Электроэнцефалография. У большинства пациентов регистрируются диффузные интериктальные симметричные эпилептогенные разряды, иктальные высокоамплитудные спайки. В ряде случаев для выявления эпиактивности требуется суточный ЭЭГ-мониторинг, проведение провокационных проб (ЭЭГ при вспышках света, гипервентиляции, резких звуковых сигналах). Результаты исследований оцениваются нейрофизиологом, эпилептологом.
- Нейровизуализация. До закрытия родничков осуществляется путём нейросонографии, у детей старше года — при помощи МРТ головного мозга. Взрослым может проводиться МСКТ. Морфологические изменения церебральных тканей характерны для симптоматических МЭ.
- Лабораторные исследования. Производятся при подозрении на наличие обменных расстройств. Включают биохимический анализ крови и мочи, специфические анализы.
- Консультация генетика. Сбор семейного анамнеза, составление генеалогического древа позволяют определить наследственный характер эпилепсии, установить тип наследования.
Дифференциальная диагностика осуществляется с неэпилептическим миоклонусом, отличительной особенностью которого выступает фокальный характер миоклоний, отсутствие реакции на провокацию, нормальная ЭЭГ-картина. Дифференцировка МЭ необходима также с судорожным синдромом инфекционной этиологии, фебрильными судорогами, синдромом Леннокса-Гасто, мозжечковой миоклонической диссинергией Ханта.
Лечение миоклонической эпилепсии
Терапия базируется на антиконвульсантах. Подбор фармпрепарата и дозировки осуществляется индивидуально. Препаратами выбора выступают производные вальпроевой кислоты, обладающие противоэпилептическим эффектом в равной степени в отношении миоклонических, клонико-тонических, абсансных пароксизмов. В фармакорезистентных случаях показано комбинированное лечение вальпроатами, бензодиазепинами, этосуксимидом, барбитуратами, антиконвульсантами нового поколения (топираматом, леветирацетамом). Важным моментом является исключение провоцирующих приступы факторов: резких звуков, вспышек света, эмоциональных всплесков, физических перегрузок, перегреваний.
Прогноз и профилактика
Наиболее прогностически неблагоприятна ранняя миоклоническая энцефалопатия, смертность составляет половину случаев заболевания, остальные дети являются глубокими инвалидами. Миоклоническая эпилепсия при болезни Лафоры, Крейтцфельдта-Якоба плохо поддаётся противоэпилептической терапии, сопровождается прогрессирующим интеллектуальным распадом. ДМЭМ и ЮМЭ отличаются доброкачественным течением, редко приводят к когнитивному дефициту. Более 50% случаев ДМЭ заканчиваются спонтанным выздоровлением.
МЭ не имеет специфических мер профилактики. К мероприятиям, способным предупредить рождение больного ребёнка, относятся планирование беременности, ранняя постановка на учёт, исключение неблагоприятных воздействий на плод. Ведение беременности должно включать разъяснительные беседы с женщиной по поводу необходимости охранительного режима, тератогенной опасности лекарственных средств, пагубного воздействия на будущего ребёнка вредных привычек.
Читайте также:
