
Детская эпилепсия синдром отахара

В 2001 году данный синдром был внесен в перечень болезней, для которых свойственны эпилептическая активность и эпилептиформные нарушения в электроэнцефалографии.
Эти нарушения вызывают развивающиеся ухудшения состояния мозга. Также была принята гипотеза Отахара о том, что данная болезнь в 80% ситуаций перерастает в другую – синдром Веста. А спустя еще время бывали ситуации с трансформацией болезни в синдром Леннокса-Гасто.
Синдром Отахара – это начальная стадия эпилептической энцефалопатии, которая выявляется у детей в первые 3 месяца после рождения. Для болезни свойственны острые приступы, которые появляются на протяжении 10-дневной жизни ребенка, в редких случаях после появления на свет.
Нарушения в метаболизме младенца говорят о семейных заболеваниях. Проявляется обостренно на фоне идеального здоровья.
Причины и этиология болезни
Самой популярной предпосылкой считаются изъяны формирования головного мозга – унилатеральная мегалэнцефалия, порэнцефалия и другие. Иногда в качестве источника могут выступать картировочные нарушения, сбои связанные с обменом веществ.
В персональных изучениях Отахар рассмотрел 10 ситуаций. В результате у 2 присутствовала киста в полушариях мозга — порэнцефалия, 
также у 2 синдром Айкарди и подострая смешанная энцефалопатия – дистрофическое изменение тканей мозга, что и вызывает нарушения его функций. У других 6 детей происхождение болезни не выявлено.
В еще одном обследовании из 11 младенцев, у 1-го была асфиксия при рождении, у 1-го с врожденной патологией, важную ценность в развитии и распространении которого играет генетика (агенезия мозолистого тела).
У 1-го некетоновая гиперглицинемия, у других основания болезни не обнаружены. Только 1 был с подобными симптомами эпилепсии как и у его родственников.
Schlumberger в своем эксперименте в 8 из 8 ситуаций поставил диагнозы, связанные с пороками головного мозга. 6 из которых – унилатеральная мегалэнцефалия и один синдром Айкарди. Унилатеральная мегалэнцефалия как причина встречалась еще в исследовательских работах Martin, Ohtsuka.
В 1995 году в статье, посвященной детской эпилепсии, было написано, что источниками синдрома Отахара считается мозговая мальформация.

В итоге после множества исследований было принято общее мнение, что структурные нарушения полушарий мозга — провокаторы заболевания.
Патофизиология
Синдромы Отахара, Веста и Леннокса-Гасто очень тесно связаны между собой, принято, что эти синдромы показывают возрастные реакции мозга в разный период его формирования.
С этим видом эпилепсии приходилось сталкиваться еще больным с полушарными или очаговыми мальформациями, порождающие тяжелые частичные припадки, которые предшествуют, но в основном следуют сразу за синдромом Отахара.
Клинические проявления
В 2002 году Aicardi и Ohtahara представили следующие главные особенности заболевания Отахара:
- болеют, как правило, младенцы сразу после рождения или в 10-дневном возрасте;
- разные типы приступов, главный – возбудительный спазм, припадки с чрезмерным напряжением мышц могут проявляться как днем, так и ночью;
- тяжелое замедление в психотропном формировании, зачастую заканчивается смертью еще в младенческом возрасте;
- возможна трансформация болезни в другие синдрома;
- в основном, все случаи связаны с нарушениями мозга.
Для синдрома Отахара свойственно прогрессирующее ухудшение состояния здоровья с учащением количества приступов, видимое замедление в психомоторном формировании организма. В основном, малыши с таким диагнозом остаются инвалидами.
Для заболевания характерны разные виды приступов. Как правило, возбудительные спазмы, которые могут распространяться по всему организму, симметричные и латерализованные относительно полушарий мозга. Средняя протяженность приступа в среднем 10 секунд, с интервалом 10-15 секунд. Также есть небольшая вероятность в проявлении других припадков.
Из-за заболевания младенцы мало активничают, проявляется гипотония. В синдром Веста болезнь может трансформировать на 2-6 месяце жизни детей, по итогам исследований процент перехода составляет около 75%. В дальнейшем заболевание имеет все шансы перерасти в синдром Леннокса-Гасто.

Диагностические процедуры
Нейровизуализация – совокупность способов, при помощи которых можно визуализировать строение, функции и биохимические свойства мозга. Данные методы необходимы для выяснения причин и назначения лечебных терапий. Как правило, при этом обнаруживают значительные отклонения от нормы и мальформации.

Если же результаты нейровизуализации в норме, проводиться метаболический скрининг.
Если медленная спайк-волновая активность, то это свойственно для синдрома Леннокса-Гасто. Во всех остальных ситуациях перерастает в тяжелую парциальную эпилепсию с высокой активностью нервных клеток в одном из полушарий.
Так как главная причина заболевания – нарушения головного мозга, то необходимо провести проверку способом нейровизуализации. Все структурные изменения можно увидеть с помощью МРТ и КТ.
При нормальных результатах нейровизуализации назначаются диагностика метаболизма. Некоторые расстройства в процессе обмена веществ оргазма могут привести к поражениям полушарий головного мозга.
Эффективность терапии практически нулевая

Противоэпилептическое лекарственное средство Фенобарбитал, также известно под маркой Люминал, способно понизить количество припадков, но антиконвульсанты не в состоянии прекратить замедление психомоторного формирования.
Ни в одном из известных случаев не было положительной реакции на лечение адренокортикотропным гормоном и антагонистов кальция. В 2001 году Fusco продемонстрировал благоприятную терапию витамином B6.
Ohno представил ситуацию с хорошим результатом на способ лечения Зонизамидом. В вариантах с гемимегалэнцефалией или кортикальной дисплазией могут помочь нейрохирургические вмешательства.
К сожалению, в настоящее время не существует действенного медикаментозного лечения болезни, половина пациентов умирает еще в младенческом возрасте от нескольких недель до месяца, у остальных развивается устойчивый неврологический и психологический недостаток.
Часто приступы при синдроме Отахара неустранимы и не поддаются лечению антиэпилептическими лекарственными средствами. Со временем возможна трансформация в другие болезни.
Если переход не был осуществлен, то психомоторное развитие будет лучше. Однако прогноз неблагоприятный, в большинстве случаев наблюдался летальный исход.
Federico Vigevano
История и терминология
В 2001г специальная отдел ILAE по классификации и терминологии предложила внести эту форму в список эпилептических энцефалопатий (Engel 2001) – заболевания, при которых не только эпилептическая активность, но и эпилептиформные изменения на ЭЭГ сами по себе могут вызывать прогрессирующие нарушения мозга. В этой группе находятся также ранняя миоклоническая энцефалопатия, синдром Веста и синдром Леннокса-Гасто. Таким образом, была признана гипотеза Отахара, что эта форма является самой ранней из возраст-зависимых эпилептических энцефалопатий, при этом в 75% случаев она затем переходит в синдром Веста, а в более позднем возрасте в некоторых случаях – в синдром Леннокса-Гасто
В 2002 г Aicardi и Ohtahara описали следующие основные характеристики синдрома (Aicardi and Ohtahara 2002):
Приступы при синдроме Отахара в большинстве описанных случаев появляются в течение 10 первых дней жизни, порой сразу после рождения (Ohtahara 1984; Clarke et al 1987). Дебютируют остро на фоне полного здоровья. Du Plessis расширил дебют заболевания до пренатального периода с внутриматочными приступами (du Plessis et al 1993). Для заболевания характерно прогрессирующее ухудшение состояния с увеличением частоты приступов и выраженной задержкой психомоторного развития Дети обычно остаются глубокими инвалидами. В описанных случаях мальчики страдают чаще девочек (Clarke et al 1987).
При синдроме Отахара встречаются различные виды приступов. Чаще всего это тонические спазмы, которые могут быть как генерализованными и симметричными, так и латерализованными. Тонические спазмы могут быть одиночными или иметь кластерное течение, возникают в бодрствовании и во сне. Продолжительность спазма около 10 секунд, интервал между спазмами в одной серии (кластере) – от 9 до 15 секунд. В 1/3 случаев встречались другие типы приступов – парциальные моторные или гемиконвульсии (Yamatogi and Ohtahara 2002). Реже встречаются массивные или сегментарные миоклонии, хаотические миоклонии нехарактерны для данного синдрома. В более позднем возрасте могут встречаться генерализованные тонико-клонические приступы.
Вскоре после дебюта заболевания дети становятся менее активными, возникает гипотония. Их психомоторное развитие останавливается, и появляется неврологический дефицит в виде спастической диплегии, гемиплегии, тетраплегии, атаксии или дистонии.
На 2-6 месяце жизни заболевание может трансформироваться в синдром Веста. По результатам наблюдения в серии пациентов такой переход произошел в 12 из 16 случаев (75%) (Yamatogi and Ohtahara 2002), с основными проявлениями в виде спазмов и гипсаритмии на ЭЭГ. В дальнейшем в 2 случаях из 12-ти синдром Веста перешел в синдром Леннокса-Гасто, с его характерной медленной спайк-волновой активностью на ЭЭГ и малыми моторными приступами.
В другом исследовании из 11 случаев, у 1 ребенка имелась асфиксия во время родов, 1 с агенезией мозолистого тела, у 1 некетоновая гиперглицинемия, в остальных 8 случаях причина не установлена (Clarke et al 1987). Только один из 8 последних случаев был отягощен семейным анамнезом по эпилепсии.
В исследовании Schlumberger у всех 8 младенцев были диагностированы пороки развития головного мозга, включая 6 случаев гемимегалэнцефалии, 1 синдром Айкарди, 1 случай дентато-оливарной дисплазии (Schlumberger et al 1992). Гемимегалэнцефалия встречалась также в исследованиях Martin, Ohtsuka, и в 2 из 8 случаях, описанных Fusco. (Martin et al 1981; Ohtsuka et al 1999; Fusco et al 2001).
В обзорной статье, посвященной злокачественным эпилепсиям детства, церебральные мальформации отмечались в качестве общей причины развития синдрома Отахара (Renier 1995). Мураками с коллегами занимались исследованием этого же вопроса (Murakami et al 1993). В нескольких исследованиях обсуждался вопрос о роли кортикальной дисгенезии в развитии ранней младенческой энцефалопатии (du Plessis et al 1993; Ogihara et al 1993; Spreafico et al 1993; Tominaga et al 1993).
Robain и Dulac представили случай с дисплазией оливарного зубчатого ядра (Robain and Dulac 1992). Описан случай диффузного нарушения церебральной миграции в сочетании с отсутствием ГАМК в спинномозговой жидкости (Miller et al 1998).
Метаболические расстройства встречались редко. В добавлении к вышеупомянутому случаю с некетонической гиперглицинемией (Clarke et al 1987), следует отметить описание случая с энцефалопатией Лея (Tatsuno et al 1984), случай с дефицитом цитохром С-оксидазы (Williams et al 1998), а также с дефицитом пиридоксина и карнитин-пальмитоилтрансферазы (Fusco et al 2001).
Несмотря на различие патоморфологических проявлений, общепринятая точка зрения сводится к тому, что синдром является следствием пороков развития головного мозга.
Патогенез и патофизиология
Вследствие тесной взаимосвязи между синдромами Отахара, Веста и Леннокса-Гасто, считается, что все они представляют возраст-зависимые реакции мозга в ходе различных стадий развития на гетерогенные, неспецифические экзогенные факторы (Ohtahara 1984). Эта форма эпилепсии встречается также у пациентов с полушарными или фокальными мальформациями, вызывающими катастрофические парциальные приступы, которые могут предшествовать, но чаще следуют после синдрома Отахара (Schlumberger et al 1992).
Относительно точные значения распространенности и заболеваемости синдрома Отахара неизвестны
Что касается синдрома Веста, то дифференциальный диагноз основывается на разнице в возрасте дебюта (раньше при синдроме Отахара), интериктальной ЭЭГ (гипсаритмия не встречается при синдроме Отахара), и типе приступов - ЭЭГ-паттерн спазмов при синдроме Веста существенно отличаются от тонических спазмов при синдроме Отахара (Fusco and Vigevano 1993).
Donat представил обзор возраст-зависимых эпилептических энцефалопатий и определил место синдрома Отахара или ранней младенческой энцефалопатии в схеме взаимоотношений между ранней миоклонической энцефалопатией, синдромом Веста и синдромом Леннокса-Гасто – все они перекрывают друг друга, что не удивительно в виду ограниченных возможностей мозга младенца (Donat 1992).
Приступы при синдроме Отахара часто некупируемые и не поддаются лечению антиэпилептическими препаратами. Как правило, имеются тяжелые нарушения психомоторного развития. Со временем, заболевание может перейти в синдром Веста или парциальную эпилепсию. Психомоторное развитие несколько лучше, если не происходит трансформации в синдром Веста и синдром Леннокса-Гасто (Clarke et al 1987). В половине описанных случаев наблюдался летальный исход в младенчестве или детском возрасте.
Фенобарбитал может уменьшить частоту приступов, но в целом антиконвульсанты мало помогают в контроле над приступами и не могут остановить нарушение психомоторного развития. Ни в одном из описанных случаев не было положительного ответа на терапию АКТГ (Clarke et al 1987). Ohno описал случай с эффектом на терапию зонизамидом (Ohno et al 2000), также Fusco представил случай с положительным ответом на лечение витамином В6 (Fusco et al 2001). В случаях с гемимегалэнцефалией или кортикальной дисплазией может помочь нейрохирургическое лечение – гемисферэктомия (Pedespan et al 1995) или фокальная резекция (Komaki et al 1999).
Список литературы
Aicardi J, Ohtahara S. Severe neonatal epilepsies with suppression-burst pattern. In: Roger J, Bureau M, Dravet CH, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3rd ed. London: John Libbey & Company Ltd, 2002:33-44.
Clarke M, Gill J, Noronha M, McKinlay I. Early infantile epileptic encephalopathy with suppression burst: Ohtahara syndrome. Dev Med Child Neurol 1987;29:520-8.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for classification of epilepsies and epileptic syndromes. Epilepsia 1985;26:268-78.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
Donat JF. The age-dependent epileptic encephalopathies. J Child Neurol 1992;7:7-21.
du Plessis AJ, Kaufmann WE, Kupsky WJ. Intra-uterine onset myoclonic encephalopathy associated with cerebral cortical dysgenesis. J Child Neurol 1993;8:164-70.
Engel J. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on classification and terminology. Epilepsia 2001;42:796-803.
Fusco L, Pachatz C, Di Capua M, Vigevano F. Video-EEG aspects of early-infantile epileptic encephalopathy with suppression-bursts (Ohtahara syndrome). Brain Dev 2001;23:708-14.
Fusco L, Vigevano F. Ictal clinical electroencephalographic findings of spasms in West syndrome. Epilepsia 1993;34(4):671-8.
Komaki H, Sugai K, Sasaki K, et al. Surgical treatment of a case of early infantile epileptic encephalopathy with suppression-bursts associated with focal cortical dysplasia. Epilepsia 1999;40:365-9.
Martin HJ, Deroubaix-Tella P, Thelliez PH. Encephalopathie epileptique neonatale a bouffees periodiques. Rev EEG Neurophysiol 1981;11:397-403.
Miller SP, Dilenge ME, Meagher-Villemure K, O'Gorman AM, Shevell MI. Infantile epileptic encephalopathy (Ohtahara syndrome) and migrational disorder. Pediatr Neurol 1998;19(1):50-4.
Murakami N, Ohtsuka Y, Ohtahara S. Early infantile epileptic syndromes with suppression-bursts: early myoclonic encephalopathy vs. Ohtahara syndrome. Jpn J Psychiatry Neurol 1993;47:197-200.
Ogihara M, Kinoue K, Takamiya H, et al. A case of early infantile epileptic encephalopathy (EIEE) with anatomical cerebral asymmetry and myoclonus. Brain Dev 1993;15:133-9.
Ohno M, Shimotsuji Y, Abe J, Shimada M, Tamiya H. Zonisamide treatment of early infantile epileptic encephalopathy. Pediatr Neurol 2000;23:341-4.
Ohtahara S. Clinico-electrical delineation of epileptic encephalopathies in childhood. Asian Med J 1978;21:499-509.
Ohtahara S. Seizure disorders in infancy and childhood. Brain Dev 1984;6:509-19.
Ohtahara S, Ishida T, Oka E, Yamatogy Y, Inoue H. On the specific age-dependent epileptic syndromes: the early-infantile epileptic encephalopathy with suppression-burst. No To Hattatsu 1976;8:270-80.
Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E. The early-infantile epileptic encephalopathy with suppression-burst: developmental aspects. Brain Dev 1987;9:371-6.
Ohtsuka Y, Ohno S, Oka E. Electroclinical characteristics of hemimegalencephaly. Pediatr Neurol 1999;20:390-3.
Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neuro 2003;20(6):398-407.
Pedespan JM, Loiseau H, Vital A, et al. Surgical treatment of an early epileptic encephalopathy with suppression-bursts and focal cortical dysplasia. Epilepsia 1995;36:37-40.
Renier WO. The malignant epilepsies of childhood and adolescence. In: Aldenkamp AP, Dreifuss FE, Reiner WO, Suumeijer PBM, editors. Epilepsy in children and adolescence. Boca Raton: CRC Press, 1995.
Robain O, Dulac O. Early epileptic encephalopathy with suppression bursts and olivary-dentate dysplasia. Neuropediatrics 1992;23(3):162-4.
Schlumberger E, Dulac O, Plouin P. Early-infantile epileptic syndrome(s) with suppression-burst: nosological considerations. In: Roger J, Bureau M, Dravet CH, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood, and adolescence. London: John Libbey & Company Ltd; 1992:35-42.
Spreafico R, Angelini L, Binelli S, et al. Burst suppression and impairment of neocortical ontogenesis: electro-clinical and neuropathologic findings in two infants with early myoclonic encephalopathy. Epilepsia 1993;34:800-8.
Tatsuno M, Hayashi M, Iwamoto H, et al. [Autopsy case of Leigh's encephalopathy with wide lesions in central nervous system and early infantile epileptic encephalopathy with burst suppression] [Japanese]. No To Hattatsu 1984;16:68-75.
Tharp BR. Neonatal seizures and syndromes. Epilepsia 2002;43:2-10.
Tominaga I, Kaihou M, Kimura T, et al. Early infantile epileptic encephalopathy (Ohtahara syndrome) with poly-microgyria. Rev Neurol (Paris) 1993;149(10):532-5.
Williams AN, Gray RG, Poulton K, et al. A case of Ohtahara syndrome with cytochrome oxidase deficiency. Dev Med Child Neurol 1998;40:568-70.
Yamatogi Y, Ohtahara S. Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev 2002;24:13-23.
Синдром Отахара в 2001 году был включен в список заболеваний, которые характеризуются повышенной эпилептической активностью, а также эпилептиформными нарушениями в показателях электроэнцефалограммы. Подобные нарушения провоцируют прогрессирующее ухудшение в работе головного мозга. В том же 2001 году была принята одноименная гипотеза, предполагающая, что в подавляющем большинстве случаев наблюдается синдром Отахара с трансформацией в синдром Веста. Также встречались случаи, когда в дальнейшем патология перерастала в синдром Леннокса-Гасто.
Описание
Синдром Марканда-Блюме-Отахара представляет собой начальный этап развития энцефалопатии эпилептического типа, которая возникает у новорожденных детей в течение первых месяцев жизни. Патология проявляется острыми приступами, которые прогрессируют на протяжении 10 дней жизни ребенка. В некоторых случаях синдром может проявиться сразу после появления ребенка на свет. Генетические заболевания могут стать причиной развития метаболических нарушений, что в итоге приводит к проявлению синдрома в острой форме на фоне хорошего состояния здоровья.
Причины
Медики склонны считать, что наиболее вероятной причиной развития синдрома Отахара у детей являются нарушения в формировании головного мозга, такие как порэнцефалия, мегалэнцефалия унилатерального типа и т.д. В некоторых случаях к патологии приводят сбои в обменных процессах, такие как картировочные нарушения.
Для персонифицированного исследования Отахар рассмотрел десять случаев. В результате удалось определить, что у двоих пациентов была в наличии киста в одном из полушарий мозга, что характеризуется как порэнцефалия. Еще у двоих пациентов был выявлен синдром Айкарди, а также энцефалопатия подострого смешанного типа. Это привело к изменениям мозговых тканей дистрофического характера и как следствие к нарушению функций головного мозга. У остальных 6 пациентов определить причины синдрома Отахара не удалось.
В ходе другого обследования рассматривались 11 новорожденных детей. Один из них испытал асфиксию во время родов, у второго была выявлена патология врожденного типа, развитие и распространение которой было обусловлено нарушениями на генетическом уровне. Еще у одного ребенка была обнаружена гиперглицинемия некетонового типа, у остальных детей выявить причину развития синдрома не удалось. И только у одного ребенка эпилептические приступы были подобны патологии, выявленной у близких родственников.
Шлумбергер также проводил эксперимент с участием 8 детей. У всех были выявлены пороки головного мозга. При этом 6 детей страдали от мегалэнцефалии унилатерального типа, а в одном случае наблюдался синдром Айкарди.
Мальформация
Еще одно предположение о причинах развития патологии Отахара было высказано в статье 1995 года, описывающей эпилепсию в детском возрасте. В данной статье говорилось о мальформации как первопричине появления синдрома. Мальформация представляет собой любое отклонение от нормы в физическом развитии, в результате которого происходят существенные нарушения в работе и строении мозга.
Таким образом, врожденные или полученные травмы головного мозга или любые другие заболевания органа могут привести к развитию синдрома у новорожденных детей. Реже встречаются случаи, когда провокатором патологии становились нарушения в обменных процессах. В итоге, на основании собранной в ходе исследований информации было принято общее мнение, что провокаторами патологии являются нарушения в структуре мозговых полушарий.
Симптомы
Основными особенностями патологии, согласно представленной Айкарди и Отахара информации, являются:
- Болезнь характерна для детей непосредственно после рождения или начиная с десятидневного возраста.
- Типы приступов могут различаться, однако наиболее распространенный – возбудительный спазм, когда происходит перенапряжение мышц. Проявляются спазмы как в дневное, так и в ночное время.
- Аномальное замедление психотропного формирования. Довольно часто заканчивается смертью ребенка в новорожденном возрасте.
- Переход синдрома в иные заболевания.
- В подавляющем большинстве случаев причиной синдрома являются нарушения в работе головного мозга.

Прогрессирующее ухудшение состояния
Синдром Отахара характеризуется прогрессирующим ухудшением состояния пациента. Приступы при этом становятся чаще с течением времени, а психомоторное развитие значительно замедляется. Дети с подобным диагнозом остаются инвалидами. Приступы могут быть как симметричными, так и латерализованными по отношению к мозговым полушариям. На фоне синдрома могут проявляться и другие разновидности припадков, не только возбудительные спазмы. Продолжительность припадка составляет 10 секунд, интервалы между приступами примерно равны 10-15 секундам.
Дети, страдающие от синдрома Отахара, малоактивны, довольно часто болезнь сопровождается гипотонией. Трансформация в синдром Веста происходит в среднем спустя 2-6 месяцев после рождения. Данный переход происходит в каждом третьем случае из четырех. В дальнейшем велика вероятность перехода патологии в синдром Леннокса-Гасто.
Диагностика
Главным диагностическим методом для выявления патологии Отахара является нейровизуализация. Это совокупность различных методик, которые дают возможность получить изображение строения, функций и свойств мозга с биохимической точки зрения. Применение этих методов позволяет выявить причины развития синдрома и назначить корректное лечение.
Нейровизуализация помогает обнаружить существенные отклонения в работе головного мозга, а также мальформации. В случае, если эти методы обнаруживают нормальные показатели, проводится так называемый метаболический скрининг. Этот метод показывает наличие нарушений в обменных процессах, что также способно стать причиной синдрома Отахара.

Интериктальная электроэнцефалография
В иных случаях патология Отахара трансформируется в парциальную разновидность эпилепсии, для которой характерна повышенная активность мозговых клеток в одном из полушарий.
Нейровизуализация предполагает проведение МРТ и КТ головы. Посредством этих исследований удается визуализировать все изменения в структуре. Фото детей с синдромом Отахара представлено ниже.
Лечение

Прогноз
На сегодняшний день, к несчастью, отсутствует действенная схема лечения синдрома. Более половины пациентов с таким диагнозом умирают в первый месяц жизни. Те, кому удалось выжить, страдают от устойчивой психологической и неврологической недоразвитости. Встречаются случаи, когда не удается даже купировать эпилептические припадки.

В некоторых случаях синдром переходит в другие заболевания. Психомоторное развитие при этом нормализуется, однако, прогноз все равно неблагоприятный.
Мы рассмотрели основные причины возникновения синдрома Отахара.
58 713 €)
4 568,97 €
$1 332,72
НУЖНО : 72 535,9 € + сопутствующие расходы
ОСТАЛОСЬ СОБРАТЬ: 8 301,9 €
ОПЛАТИТЬ ДО: 28.10.10
ОСТАЛОСЬ 5 ДНЕЙ.
Маленькой Саше всего год. Она больная, детская эпилепсия, самый страшный диагноз - синдром Отахара, что означает: противосудорожные препараты помочь не могут. Помочь крохе может только операция в Германии - шанс на жизнь.
Детки с подобным заболеванием живут до двух лет и умирают в муках, с мучительной болью. Ей очень хочется жить, научиться узнавать маму, улыбаться, ходить. просто жить.
дата операции - декабрь 2010 года, госпитализация в ноябре 1010 года. В настоящий момент у Саши ухудшение состояние здоровья - приступы эпилепсии каждый день, ребенок питается 3 месяца через зонд. Несколько дней подряд держится температура 40. Ребенок находится в крайне тяжелом состоянии,в бреду, постоянно стонет, в данный момент решается вопрос госпитализации для снятия этих симптомов - но излечить причину может лишь операция.
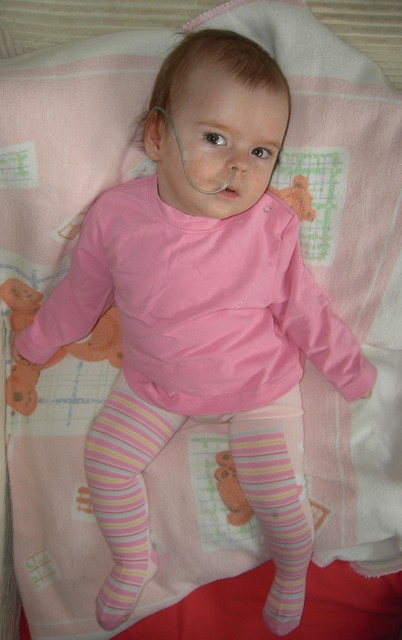
Здравствуйте! Меня зовут Лиза, проживаю в г. Зеленоград, Москва.
27 сентября 2009 года у меня родилась дочь Саша. Роды прошли нормально, получили 8-9 Апгар.. А через 2 часа после рождения началиь судороги, через каждые пару часов, потом все чаще.. Из роддома Сашку перевели в больницу для недоношенных деток (мол там больше возможностей для обследования и тд).
К сожалению, находится там с ней было можно только несколько часов в день, и каждый раз меня просто выгоняли оттуда. Обследования и осмотры специалистов ничего не давали - природу судорог не помогли определить ни анализы крови, ни пункции спинномозговой жидкости. ЭЭГ показывала наличие очагов эпи-активности, причину которых никто назвать не мог. В очередной раз мне сказали, что здесь нам помочь ничем не могут и переведут в специализированный исследовательский центр. Так мы оказались в Солнцево. Здесь Саше сделали первое МРТ, которое показало врожденный порок мозга, в том числе и гемимегалэнцефалию (одна половинка мозга больше другой). Был поставлен диагноз - синдром Отахара, один из самых страшных, злокачественных видов младенческой эпилепсии, который не поддается лечению. Мне сказали, что мой ребенок не будет ходить, говорить. и вообще - большой процент таких деток умирает в раннем возрасте. Невозможно описать, что чувствуешь, услышав такое. не дай Бог кому. Тем не менее врачи начали подбирать нам противосудорожную терапию, хотя предупредили- эффект будет кратковременным. Рассказали также, что единственный наш шанс, это если порок окажется операбельным. Приступы к тому времени уже имели серийный характер - до 150-200 за серию, по 10 серий в сутки. В этом центре мы провели почти месяц, затем, получив положительную динамику на клоназепаме, нас выписали домой.
Первые несколько месяцев после выписки из Центра все было отлично - Сашка развивалась, начала гулить, поднимать голову и даже топать ножками. Мы были очень счастливы и бегом наверстывали упущенное время.
Но через 2,5 месяца приступы возобновились, сначала однократно, потом по нескольку раз. После одного-двух развитие обнулялось и, если какое-то время было спокойно, она начинала учиться заново, до нового приступа. Но, конечно, и за 2 недели, мы не успевали даже начать снова как следует держать голову. Количество приступов росло, а промежутки между ними сокращались - от нескольких недель в начале, до нескольких часов через 2 месяца. Она замолчала, перестала улыбаться, фиксировать взгляд и не хотела двигаться, только тихонько скулила, если было очень больно. Особенностью наших эпилептических приступов было еще и то, что во время них она находилась в сознании, поэтому очень пугалась и страшно кричала.
Положение осложнилось тяжелым обструктивным бронхитом, схваченым однажды в больнице и не покидавшим нас потом еще долгие месяцы. Почти полное отсутствие иммунитета (что-то там с тимусом) и лежачий образ жизни не давали возможности покинуть инфекционное отделение и заняться неврологией. Повышение доз конвулекса и клоназепама не давало эффекта, и постепенно число приступов выросло до беспрерывного (статусного) их течения.
К июню их было уже несколько видов. Попытки снять их наркозом или уколом реланиума не имели результата, поэтому врачами было решено не предпринимать ничего до тех пор, пока она сама выходит из приступа и успевает восстановить дыхание до следующего. Также появился бульбарный синдром (частичный паралич глотки и соответственно - потеря глотательных и сосательных рефлексов), поэтому мы начали кормиться через зонд.
В таком виде нас уже перевели в неврологию. Еще несколько недель врачи отделения подбирали комбинации противосудорожных препаратов, в итоге положительный эффект был достигнут на люминале (фенобарбитал) в очень высокой дозировке, и в конце июня этого года мы выписались.
К сожалению, очень скоро приступы возобновились, сейчас они бывают в среднем раз в неделю, но после каждого следующего она все дольше восстанавливается. Общее развитие остается на начальном этапе. До сих пор не улыбается. Из-за высокого риска асперационной пневмонии (слюна и пища постоянно попадают в дыхательные пути, их все время необходимо отсасывать оттуда) постоянно пьем антибиотики.
Почему-то, врачи в Солнцево еще тогда, в ноябре, не дали нам заключение о необходимости операции как единственной возможности на улучшение. Имея его, мы могли обратиться в клинику уже в тот период ремиссии (ноябрь-январь 2009). Бумагу именно с такой формулировкой мы получили только в нашу последнюю госпитализацию в Морозовской больнице (июнь 2010), когда ребенок уже месяц находился в тяжелом состоянии.
В ходе уточняющих обследований выяснилось, что операция будет заключаться, скорее всего, в удалении поврежденного полушария мозга полностью. Как правило, чем меньше возраст, тем легче происходит адаптация после операции и оставшаяся часть мозга должна взять на себя функции удаленной. И, наверно, после длительной реабилитации, Сашка сможет ходить, говорить..
Мы сразу связались с рекомендованной нам клиникой в Германии и, отправив все необходимые мед.документы, вскоре получили ответ: Профессор Хольтхаузен согласился с необходимостью срочной операции и взялся ее сделать. Нам была назначена дата приезда на операцию - в феврале будущего года (2011). Стоимость операции и пребывания в клинике составляет 80 тысяч евро. Такую сумму самим собрать за пару лет трудно , а за несколько месяцев - нереально.. Но и откладывать операцию нельзя, состояни ребенка ухудшается день ото дня. Сколько еще протянем в таком виде без осложнений неизвестно, но мы очень стараемся дотянуть до февраля.
В настоящий момент ребенок уже 3 месяца получает питание только через зонд, количество приступо - практически ежедневно. Третий день держится температура 40, ничем не сбивается,врачи разводят руками. Девочка находится дома.
Мы знаем,что у нас есть шанс на жизнь. Очень хочется верить в то, что нам смогут помочь осуществить нашу мечту. С миру по нитке - так, может, мы сможем выткать одеяло жизни для маленькой девочки.
СВИДЕТЕЛЬСТВО О РОЖДЕНИИ 
ВЫПИСКА ИЗ ИСТОРИИ БОЛЕЗНИ И ДАННЫЕ ОБСЛЕДОВАНИЙ 





СЧЕТ ИЗ ГЕРМАНИИ НА ПРОВЕДЕНИЕ ОПЕРАЦИИ 

СЧЕТА ДЛЯ ОКАЗАНИЯ МАТЕРИАЛЬНОЙ ПОМОЩИ
ДЛЯ ПОЧТОВОГО ПЕРЕВОДА
124498, г. Москва, г. Зеленоград, Центральный проспект, корпус 407, кв. 76
Получатель: Гаврилова Елизавета Леонидовна
ВЕБМАНИ
R352058071023 рубли
Z149006349112 доллары
E157294280618 евро
Для переводов в Германии и Европе через немецкий благотворительный фонд "Еin recht auf leben"
Ein Recht auf Leben e.V.
Konto 6200102500
BLZ 47260121 Volksbank Padeborn.
IBAN: DE53472601216200102500
BIC: DGPBDE3MXXX
Не забудьте указать, для кого платеж (VWZ)
VWZ: Aleksandra Gavrilova
PayPal:
ein-recht-auf-leben@web.de
Не забудьте указать, для кого платеж (VWZ)
VWZ: Aleksandra Gavrilova
В случае, если у Вас такого поля нет, то напишите мне в ЛС ваш аккаунт PayPal, сумму и дату перевода, деньги поступят на счет Саши.
e-mail: gavrilova.eliz@yandex.ru
+++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
КОНТАКТЫ ЛИЗЫ
Адрес:
124498, г. Москва, г. Зеленоград, Центральный проспект, корпус 407, кв. 76
Получатель: Гаврилова Елизавета Леонидовна
Телефон
8-965-258-67-38
оператор "Билайн"
НАШИ НОВОСТИ
БОЛЬШАЯ ПРОСЬБА:
кто отвечает за форумы - уберите отовсюду, по возможности, номер телефона, на который можно отправить деньги.
СПАСИБО.
Наши действия на будущее:
На 23.10 назначена съемка ролика про Сашу
Отредактировано: Kizo в 11 апр 2011, 23:12
Присоединённые изображения

Читайте также:
