
Эпилепсия при демиелинизирующих заболеваниях

Демиелинизирующая болезнь формирует разнородную клинико-морфологическую группу, главный представитель которой – рассеянный склероз. Болезнь, в основном, поражает молодых людей, имеет прогрессивный, преимущественно, долгосрочный хронический курс. Основная морфологическая особенность – демиелинизация, возникающая в результате поражения олигодендроглии или прямого повреждения миелина.
Что это такое?
Демиелинизирующее заболевание головного мозга образуется вторично в связи с инфекциями – воспалительными, вирусными заболеваниями или вакцинацией, иногда является последствием метаболического или генетически обусловленного нарушения метаболизма миелина. Другие причины – гипоксическо-ишемическое повреждение, отравление.
Фактор риска демиелинизации головного мозга – лихорадочные заболевания, симптомы болезни могут быть весьма серьезными. При этих заболеваниях происходит постепенное разрушение миелина (= липопротеин, формирующийся с помощью белков и жирных веществ, задача которого заключается в ускорении проводимости кислорода). Демиелинизирующее заболевание протекает относительно долго, постепенно улучшается. Демиелинизирующие заболевания происходят в результате разрушения миелина, как в ЦНС, так и в ПНС.
Наиболее распространенные заболевания демиелинизирующие болезни:
- рассеянный склероз;
- диффузный склероз, известный также как болезнь Шильдера;
- полинейропатия, затрагивающая ПНС;
- амиотрофический латеральный склероз;
- ОДЭМ = острый диссеминизированный энцефаломиелит.

В соответствии с МКБ-10 демиелинизирующие болезни определяются по следующему коду: G00-G99 – болезни нервной системы → G35-G37 – демиелинизирующие заболевания ЦНС.
Рассеянный склероз
Sclerosis multiplex cerebrospinalis (множественный цереброспинальный склероз, рассеянный склероз) – это хроническое демиелинизирующее заболевание, при котором иммунная система человека атакует центральную нервную систему. В наших географических условиях является довольно распространенной болезнью. РС затрагивает около 60-100/100 000 (и даже больше) человек, особенно людей молодых и среднего возраста. Иногда может развивается контрастно – у ребенка (МРТ показывает проявления незавершенной миелинизации) или, наоборот, в пожилом возрасте. Заболевание чаще проявляется в 20-40-летнем возрасте, затрагивает больше женщин (соотношение женщин и мужчин составляет 2:1). Точная причина демиелинизирующей болезни пока неуточненная, но в настоящее время РС считается хроническим воспалительным аутоиммунным расстройством, имеющим происхождение в нарушении клеточного иммунитета. Развитие демиелинизирующего заболевания находится под влиянием нескольких факторов – генетических, окружающей среды.
Миелинизация – это процесс формирования оболочек миелина на нервных волокнах. Миелин начинает копиться в течение пренатального периода – около 5 месяца развития, заканчивает – к 2-м годам жизни.
Рассеянный склероз непредсказуем, он проявляется индивидуально у каждого человека. При более легкой форме признаки в течение некоторого времени могут исчезнуть. В других случаях демиелинизирующая болезнь атакует быстро, оставляет постоянные последствия. Преобладающие клинические признаки зависят от местоположения очагов демиелинизации головного мозга. В начале болезни проявляется следующая клиническая картина:
- Оптический ретробульбарный неврит – одностороннее расстройство зрения, которое обычно быстро и полностью корректируется. Иногда остается центральный скотом (место на сетчатке, физиологически не реагирующее на падающий свет, также известное как слепое пятно).
- Чувствительные симптомы – в основном параэстезия (неприятное ощущение покалывания, зуда, жжения кожи без длительных эффектов) в верхних и нижних конечностях, в основном, асимметричная. Слабость или онемение одной или нескольких конечностей – первый признак этого заболевания примерно у половины пациентов. У более молодых пациентов вторичным признаком может быть невралгия тройничного нерва.
- Вестибулярный синдром (нарушение баланса вестибулярного аппарата) – в основном центральный, иногда с сильным головокружением. Нистагм (колебательное движение глазных луковиц) распространен даже без субъективных вестибулярных симптомов. Частые симптомы – это диплопия (двойное зрение) и интернуклеарная офтальмоплегия (поражение нервов шейного отдела – затылочных). Иногда РС может начаться картиной острого диссеминированного энцефалита (острого воспаления головного мозга).
- Спастические двигательные симптомы – в начале заболевания отсутствует выраженный паралич или спастичность (повышенное напряжение мышечных волокон с более или менее частым мышечным подергиванием). Симптомы сопровождаются повышенной усталостью, неопределенностью в ходьбе, слабостью, неуклюжестью. В ходе обследования обнаруживается гиперрефлексия (повышенные рефлексы) со спазматическими пирамидальными явлениями, часто – приглушение абдоминальных рефлексов.
- Расстройства мозжечка – распространенный мозговой симптом, варьирующийся по интенсивности, от легкой атаксии (нарушение координации движения) одной конечности до тяжелой атаксии ходьбы, нарушений равновесия, которые также могут быть связаны с атаксией позвоночника (нарушение равновесия ухудшается в темноте и при закрытых глазах).
- Нарушения сфинктеров, особенно мочеиспускательного, частые позывы к мочеиспусканию, которые должны быть немедленно выполнены (мочевой пузырь имеет низкую емкость, часто сокращается, когда накапливается содержимое). Позднее добавляется удержание или недержание мочи. Могут также возникнуть сексуальные расстройства (импотенция).
- Психические симптомы – аффективность, депрессивные синдромы, эйфория. Интеллект не повреждается. Распространена усталость.
Другие проявления очень разнообразны. Существуют разные варианты опасной злокачественной формы, при которой в течение нескольких лет пациент прикован в постели. Может наступить смерть. При доброкачественных формах в течение многих лет больной подвижен, имеет лишь незначительные симптомы. Тем не менее, в большинстве случаев в течение нескольких лет прогрессивно снижается динамика, в ней могут преобладать спастические и мозжечковые симптомы с тяжелой атаксией, преднамеренным тремором, комбинированными спастическо-атаксичными проявлениями.

Изменения и нарушения сосудистого генеза часто приводят к психическим расстройствам. Но, субкортикальные локализации не обязательно имеют симптомы – они могут протекать в латентном состоянии. Дисциркуляторные очаги (многоочаговое поражение) имеют выраженную симптоматику.
Диагностика
Демиелинизирующий процесс головного мозга требует проведения тщательного диагноза:
- МРТ – магнитная томограмма показывает гиперсигнальные очаги в белом веществе головного и спинного мозга на изображениях Т2 и гипосигнальные очаги – на Т1 (количество коррелирует с тяжестью заболевания);
- исследование цереброспинальной жидкости – интратекальный синтез IgG, наличие по меньшей мере 2 олигоклональных полос в щелочной части спектра, не присутствующей в сыворотке, плейоцитоз мононуклеарных клеток (до 100/3), распространенность лимфоцитов;
- исследование вызванных потенциалов (визуальных и соматосенсорных) – увеличение латентности волн;
- офтальмологическое обследование (в случае ретробульбарного неврита) – в острой стадии присутствует отек папилы, позднее – его временное замирание (проявление атрофии);
- гистология – указывает на присутствие воспалительных изменений в голове, дистрофические процессы, дегенерацию нейронов (глиоз).

Лечение
Терапия сосредоточена на влиянии на иммунореактивный процесс. К сожалению, причина рассеянного склероза не может быть устранена. Поэтому лечение направлено на улучшение здоровья пациента, смягчение его состояния. Используемые методы можно разделить на 3 группы:
- Кортикоиды – доминирующие средства в период обострения. Они вводятся в основном в период приступа, преимущественно, внутривенно в инфузиях. Применяется Метилпреднизолон. Если терапевтический эффект недостаточен, можно однократно использовать Циклофосфамид.
- В период профилактики рецидивов используются поддерживающие дозы Преднизона или других иммунодепрессантов (Азатиоприн, Циклофосфамид) для замедления дальнейшей атаки и стабилизации процесса. Этот вид лечения в основном используется на ранних стадиях у приступообразной формы. Долгосрочное применение кортикостероидов чревато рядом возможных побочных эффектов, поэтому они всегда вводятся в разовой суточной дозе через день (чередующийся метод).
- Симптоматическое лечение руководствуется клиническими проявлениями. При спастичности применяются миорелаксанты (Баклофен, Тизанидин, Тетразепам). Церебральные симптомы улучшает Физостигмин, тремор – Клоназепам, иногда в сочетании с Тримепранолом. Парестезию может облегчить Карбамазепин, Амитриптилин или гидантоины. Мелипрамин или Дитропан можно использовать против нарушения мочеиспускания. Частью лечения является хорошо подобранная реабилитация. Регулярное целенаправленное лечение важно при наличии определенных провокационных факторов и улучшения прогноза.
Диффузный склероз Шильдера
Демиелинизирующее заболевание также известно, как болезнь Шильдера. Она поражает детей и молодых людей, имеет ненаследственный характер, прогрессивное (быстро развивающееся) течение. Максимальное время выживания – 10 лет.
Основа демиелинизирующей болезни – значительный очаг разрушения миелина, по всей мозговой доле, как правило, распространяющийся по мозолистому телу (каудальное тело, ответственное за присоединение к полушариям головного мозга). Кроме того, в стволе, спинном мозге и зрительных нервах имеются многочисленные единичные небольшие демиелинизирующие поражения.

Развитие болезни Шильдера приводит к следующим проявлениям:
- гемиплегия (паралич всей левой или правой стороны тела) или квадриплегия (полный или частичный паралич всех 4-х конечностей и туловища);
- омонимичная гемианопсия (затемнение половины поля зрения);
- кортикальная слепота, глухоты;
- псевдобульбарный синдром.
Диагностика и лечение – такие же, как при рассеянном склерозе.
Полиневропатия, поражающая ПНС
Полиневропатия – это группа демиелинизирующих заболеваний периферических нервов, поражающих представителей обоих полов. В возникновении болезни участвуют влияния эндогенных и экзогенных факторов:
- воспаление;
- нарушение обмена веществ;
- токсические, иммунопатогенные эффекты;
- дефицит витаминов, питательных веществ;
- дегенеративные, паранеопластические факторы и др.
- наследственные: моторная и сенсорная невропатия;
- метаболические: сахарный диабет, уремия, печеночные расстройства, порфирия, гипотиреоз;
- дефицит питания: недостаток витаминов B12, B1, алкоголь, недоедание, мальабсорбция;
- токсичные: алкоголь, наркотики, органические промышленные вещества (бензол, толуол, акриламид, дисульфид углерода), металлы (ртуть, свинец);
- воспалительно-аутоиммунные: синдром Гийена-Барре, острые и хронические воспалительные демиелинизирующие невропатии, болезнь Лайма;
- аутоиммунные заболевания: узелковый полиартериит, ревматоидный артрит, системная красная волчанка, саркоидоз;
- паранеопластические: рак, лимфопролиферативные расстройства, миелома, гаммапатия.
Затрагиваются, в основном, длинные нервы, клинические проявления преобладают в дистальных отделах конечностей (на противоположной стороне поражения). Признаки могут быть симметричными и асимметричными. Нередко преобладают симптомы чувствительные, двигательные, вегетативные в отношении 3-х основных типов нервных волокон.
Первые симптомы включают жжение, покалывание, чувство стянутости, холода, боль, снижение чувствительности. Эти признаки обычно начинаются на стопах ног, откуда распространяются на лодыжки. Позже, когда поражения вырастают в объемах, проявления достигают области берцовой кости, появляются на пальцах рук.
Среди двигательных проявлений наиболее частые – судороги, тремор мышц (подергивание), слабость мышц, атрофия, повышенная усталость, неуверенная походка.
Расстройство вегетативных нервов вызывает нарушение потливости, кровообращения, сердечного ритма. Происходит снижение рефлексов, нарушение обоняния, поражение глубокой чувствительности и т. д.

Полиневропатия может развиваться остро и хронически. Демиелинизирующее заболевание требует полного профессионального обследования. Необходимо исследовать кровь, мочу (исключить причины диабета, расстройства печени, почек и т.д.). Проводится рентген грудной клетки, ЭМГ, исследование цереброспинальной жидкости (люмбальная пункция), обследование членов семьи, нервная и мышечная биопсия.
Лечить полинейропатию можно после определения ее причины. Основа терапии – именно устранение причинного фактора. Значительная часть лечебного процесса – поддерживающая, направленная на регенерацию периферических нервов, но не затрагивающая основные причины, не излечивающая демиелинизирующую болезнь. Неприятные проблемы устраняются антидепрессантами, седативными средствами. Для уменьшения нервного раздражения используется Карбамазепин или Фенитоин.
Латеральный амиотрофический склероз (ЛАС)
Заболевание также известно как болезнь Лу Герига или болезнь мотонейрона.
ЛАС начинается незаметно. У пациента возникают трудности с обычными действиями (письмо, застегивание пуговиц). Позже развиваются проблемы с ходьбой, мышечные судороги, трудности с глотанием. В конце больной полностью парализован, зависит от искусственного дыхания, питания. При смертельном ЛАС пациент постепенно теряет нервные клетки. В 5-10% случаев заболевание имеет наследственную форму.
Демиелинизирующее заболевание характеризуется селективным нарушением центральных и периферических моторных нейронов (моторный нейрон – нервная клетка, непосредственно иннервирующая скелетные мышцы). Речь идет о постепенном исчезновении мотонейронов спинного мозга, в области мозгового ствола, дегенерации кортикоспинального пути. Для положительного заключения в обнаружении заболевания требуется потеря около 40% моторных нервов. До тех пор болезнь не имеет никаких очевидных симптомов. К сожалению, причина расстройства пока неизвестна. Это – комбинированное поражение I и II двигательных нейронов.
Типичные проявления: мышечная атрофия, фасциоз, тендинококковая гиперрефлексия, спастические явления. Болезнь начинается около 40-50-летнего возраста (чаще – до 60 лет). Развивается неуклюжесть рук, позже добавляются трудности при ходьбе. Вскоре возникает атрофия мышц. Могут присутствовать мышечные спазмы, боли. Иногда болезнь начинается с бульбарного синдрома, трудностей с речью, глотанием.
Лечения демиелинизирующего недуга не существует, терапия только симптоматичная, но не очень эффективная, не целенаправленная. Некоторая задержка в ходе заболевания была продемонстрирована при применении препарата Рилузол (Рилутек). Прогрессирование очень быстрое, обычно в течение 5 лет с момента начала заболевания.

ОДЭМ = острый диссеминизированный энцефаломиелит
Демиелинизирующее заболевание головного и спинного мозга, происходящее после острых вирусных инфекций (корь, краснуха и т.д.) или некоторых видов вакцинации (например, от бешенства, оспы и др.). Это, скорее, иммунологическое осложнение инфекции, чем прямое поражение ЦНС вирусом.
Клиническая картина не отличается от других энцефаломиелитов. Ликворологические данные либо нормальные, либо показывают диссоциацию цитологического белка.
Заключение
Тема демиелинизирующих болезней отражает медицинскую и социальную значимость этих недугов, подводные камни диагностики, новые тенденции в лечении в ситуации постоянного увеличения числа пациентов с такими проблемами. Речь идет о постоянной теме большинства конгрессов, поскольку демиелинизирующие расстройства – одни из самых серьезных заболеваний ЦНС, в основном, с тяжелыми последствиями, требующие специализированного подхода, не предполагающие применения народных терапевтических методов.
1. К зонам высокого риска рассеянного склероза относятся
2. В патогенезе рассеянного склероза основная роль отводится
1 – воспалительным реакциям
*2 – аутоиммунным реакциям
3 – травматическому повреждению мозга
4 – токсическому воздействию на мозг
3. Какие симптомы характерны для начального периода рассеянного склероза
*1 - нарушение зрения
2 - снижение слуха
*3 - слабость в ногах
*4 - нарушение походки
5 - эпилептические припадки
*6 - неуверенность, пошатывание при ходьбе
4. Перечислите основные системы головного и спинного мозга, страдающие при рассеянном склерозе
1 - проводники поверхностной и глубокой чувствительности
*2 - зрительный нерв и зрительный путь
*3 - пирамидная система
4 - экстрапирамидная система
*5 - мозжечковая система
6 - периферические нервы
5. Укажите, какие симптомы возникают при рассеянном склерозе
*1 - побледнение височных половин сосков зрительных нервов
*3 - интенционное дрожание, нистагм
4 - хореиформный гиперкинез
*5 - нижний спастический парапарез
*6 – императивные позывы на мочеиспускание
6. Укажите основные симптомы, характерные для рассеянного склероза
*2 - скандированная речь
3 - полиневритический тип нарушения чувствительности
*4 – повышение глубоких рефлексов
5 – гипертоно-гипокинетический синдром
*6 - атрофия дисков зрительных нервов
*7 – патологические рефлексы
*8 - отсутствие брюшных рефлексов
7. Укажите характерные для рассеянного склероза типы течения
*2 – первично прогрессирующий
*4 – вторично прогрессирующий
8. Для диагностики рассеянного склероза информативны
*1 – магнито-резонансная томография
*4 – исследование вызванных потенциалов мозга
6 – компьютерная томография
9. Укажите основные принципы патогенетического лечения рассеянного склероза
*1 – кортикостероидная терапия
*3 – иммунопревентивная терапия
4 – лучевая терапия
*6 – цитостатическая терапия
8 – противосудорожная терапия
10. Для симптоматической терапии рассеянного склероза целесообразно применение
*6 – детрузитола, десмопрессина
Укажите особенности этиологии и патогенеза острой демиелинизирующей полиневропатии (синдрома Гийена-Барре)
этиологическим фактором является экзогенная интоксикация
*отмечается аутоиммунное поражение периферических нервов и корешков
отмечается аутоиммунное поражение периферических нервов и шейного утолщения спинного мозга
*часто развивается после предшествующей острой инфекции
часто развивается на фоне хронической инфекции
отмечается компрессионно-ишемическое поражение периферических нервов
Каковы клинические проявления острой демиелинизирующей полиневропатии (синдрома Гийена-Барре)
*боли в конечностях в дебюте заболевания
полиартрит в дебюте заболевания
*периферические парезы конечностей
*белково-клеточная диссоциация в СМЖ
эозинофильный плеоцитоз в СМЖ
патологические стопные знаки
*резкое снижение сухожильных рефлексов
Какие методы лечения применяются при острой демиелинизирующей полиневропатии (синдроме Гийена-Барре)
ультрафиолетовое облучение крови
*иммуноглобулин G внутривенно
иммуноглобулин G эндолюмбально
Укажите особенности этиологии и патогенеза амиотрофического бокового склероза
этиологическим фактором является эндогенная интоксикация
этиологическим фактором является бактериальная инфекция
*отмечается прогрессирующее поражение двигательных нейронов спинного и головного мозга
отмечается прогрессирующее поражение двигательных и чувствительных нейронов спинного мозга
в патогенезе участвует механизм ишемического каскада
*в патогенезе участвует механизм глутаматной эксайтотоксичности
Каковы клинические проявления амиотрофического бокового склероза
*мышечные атрофии дистальных отделов конечностей
*высокие сухожильные рефлексы
низкие сухожильные рефлексы
*чувствительные расстройства не характерны
чувствительность нарушена по полиневритическому типу
1. Какие припадки выделяют по причине возникновения и механизму развития (этиопатогенетическая классификация Шанько Г.Г., 1990)
*7 неопределенного генеза
2. Какие виды эпилептических припадков выделяют по международной классификации 1981 г.
3. Перечислите периоды первично-генерализованного судорожного припадка
* 3 - потеря сознания
* 5 - восстановление сознания
* 6 - послеприпадочный сон
7 - постприступный период
4. Перечислите признаки простых и сложных абсансов
1 чаще в возрасте до 4 лет
*2 чаще в возрасте от 4 до 13 лет
3 чаще в возрасте после 13 лет
*6 отсутствие генерализованных конвульсий
7 выраженные вегетативные нарушения
*8 отсутствие постприпадочных нарушений
9 отсутствие нарушения сознания
5. Каковы клинические проявления и топика парциальных моторных джексоновских припадков
*1 прецентральная извилина
2 постцентральная извилина
3 насильственный поворот головы и глаз
4 сосательные, глотательные, жевательные движения
*5 клонические, тонические судороги в отдельных группах мышц в руке, ноге или лице
6. Каковы клинические проявления и топика парциальных моторных адверсивных припадков
1 прецентральная извилина
*2 средняя лобная извилина
3 сосательные, глотательные, жевательные движения
*4 насильственный поворот головы и глаз
5 клонические, тонические судороги в отдельных группах мышц в руке, ноге или лице
7. Какие клинические проявления и топика парциальных соматосенсорных джексоновских припадков
1 передняя центральная извилина
*2 постцентральная извилина
3 насильственный поворот головы и глаз
*4 парестезии, онемение в руке, ноге или лице
5 клонические, тонические судороги в отдельных группах мышц в руке, ноге или лице
8. Укажите признаки эпилептического статуса
*1 отсутствие сознания в межприступном периоде
2 длительность припадка до 10 минут
3 длительность припадка до 20 минут
*4 длительность припадка до 30 минут
5 частые припадки (до 10) в течение суток
6 частые припадки (до 30) в течение суток
7 частые припадки (до 50) в течение суток
9. Укажите основные принципы лечения эпилепсии
4 длительность эффективной терапии 1 год
*5 длительность эффективной терапии от 2 до 5 лет
6 длительность эффективной терапии постоянная
*7 постепенная отмена препарата
8 быстрая отмена препарата
10. Какие противоэпилептические препараты относятся к препаратам первого выбора
11. Укажите признаки обморока для дифференциальной диагностики с эпилепсией
*1 наличие провоцирующего фактора
2 спонтанное начало
3 кратковременные, однообразные вегето-висцеральные нарушения
*4 разнообразные, постепенно нарастающие вегето-висцеральные нарушения
5 быстрое нарушение сознания
*6 постепенное нарушение сознания
*7 быстрое восстановление сознания
8 постепенное восстановление сознания
12. Укажите признаки истерического припадка для дифференциальной диагностики с эпилепсией
*1 наличие провоцирующего фактора
2 спонтанное начало
3 нарушение сознания
*4 сознание не нарушено
5 судороги четко очерчены
*6 отсутствие четкого характера судорог
7 возможно недержание мочи и кала
*8 никогда не бывает недержание мочи и кала
9 наличие постприступного периода
*10 отсутствие постприступного периода
13. У больного снижена сила в левых конечностях, с повышением мышечного тонуса и сухожильных рефлексов, патологическими стопными рефлексами, с периодическими судорожными подергиваниями в левой руке без нарушения сознания. Где локализуется патологический очаг, назовите клинические синдромы
2 верхняя и средняя треть прецентральной извилины слева
*3 верхняя и средняя треть прецентральной извилины справа
4 центральный парапарез
*5 центральный гемипарез
6 сложные парциальные моторные припадки
*7 простые парциальные моторные припадки
8 сложные парциальные соматосенсорные припадки
9 простые парциальные соматосенсорные припадки
14. У больного периодически возникают парастезии в руке и половине лица справа, протекающие с нарушением сознания. Где локализуется патологический очаг и как называются припадки
*1 постцентральная извилина слева
2 средняя лобная извилина справа
3 средняя лобная извилина слева
4 постцентральная извилина справа
5 прецентральная извилина справа
6 прецентральная извилина слева
7 сложные парциальные моторные припадки
8 адверсивные припадки
9 простые парциальные моторные припадки
*10 сложные парциальные соматосенсорные припадки
11 простые парциальные соматосенсорные припадки
15. У больного отмечаются периодические тонико-клонические припадки с потерей сознания продолжительность около 2-3 минут с последующей вялостью и сном. Перед припадком, за 5-10 секунд, появляется насильственный поворот головы и глаз вправо. Где локализуется патологический очаг и как называются припадки
1 постцентральная извилина слева
2 средняя лобная извилина справа
*3 средняя лобная извилина слева
4 постцентральная извилина справа
5 прецентральная извилина справа
6 прецентральная извилина слева
7 эпилепсия, сложные парциальные моторные джексоновские припадки
8 эпилепсия, сложные парциальные адверсивные припадки
9 эпилепсия, простые парциальные моторные припадки
10 эпилепсия, генерализованная, тонико-клонические припадки
*11 эпилепсия, простая парциальная адверсивная, с вторично-генерализованными тонико-клоническими припадками
Последнее изменение этой страницы: 2016-04-23; Нарушение авторского права страницы
Эпилепсия — заболевание головного мозга, соответствующее любому из следующих состояний:
- Не менее двух неспровоцированных (или рефлекторных) эпилептических приступов с интервалом > 24 ч.
- Один неспровоцированный (или рефлекторный) эпилептический приступ и вероятность повторных приступов, соответствующая общему риску рецидива (³ 60 %) после двух неспровоцированных эпилептических приступов, в следующие 10 лет.
- Диагноз эпилептического синдрома.
Критерии разрешения эпилепсии включают достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом либо отсутствие эпилептических приступов в течение 10 лет у пациентов, не получавших противосудорожные препараты более 5 лет.
Классификация эпилепсии проводится после определения критериев диагностики эпилепсии (определение выше). Классификация проводится с использованием трехуровнего ранжирования — определение типа приступов, типа эпилепсии и синдрома эпилепсии. Нейроимиджинг, ЭЭГ и другие исследования, если они есть, помогают улучшить классификацию на всех трех уровнях. Где это возможно, следует установить диагноз на всех трех уровнях. Этиологию эпилепсии следует устанавливать с самого начала и на каждом этапе всего диагностического пути. Знание этиологии может способствовать оптимизации классификации и имеет важные лечебные последствия для пациента.
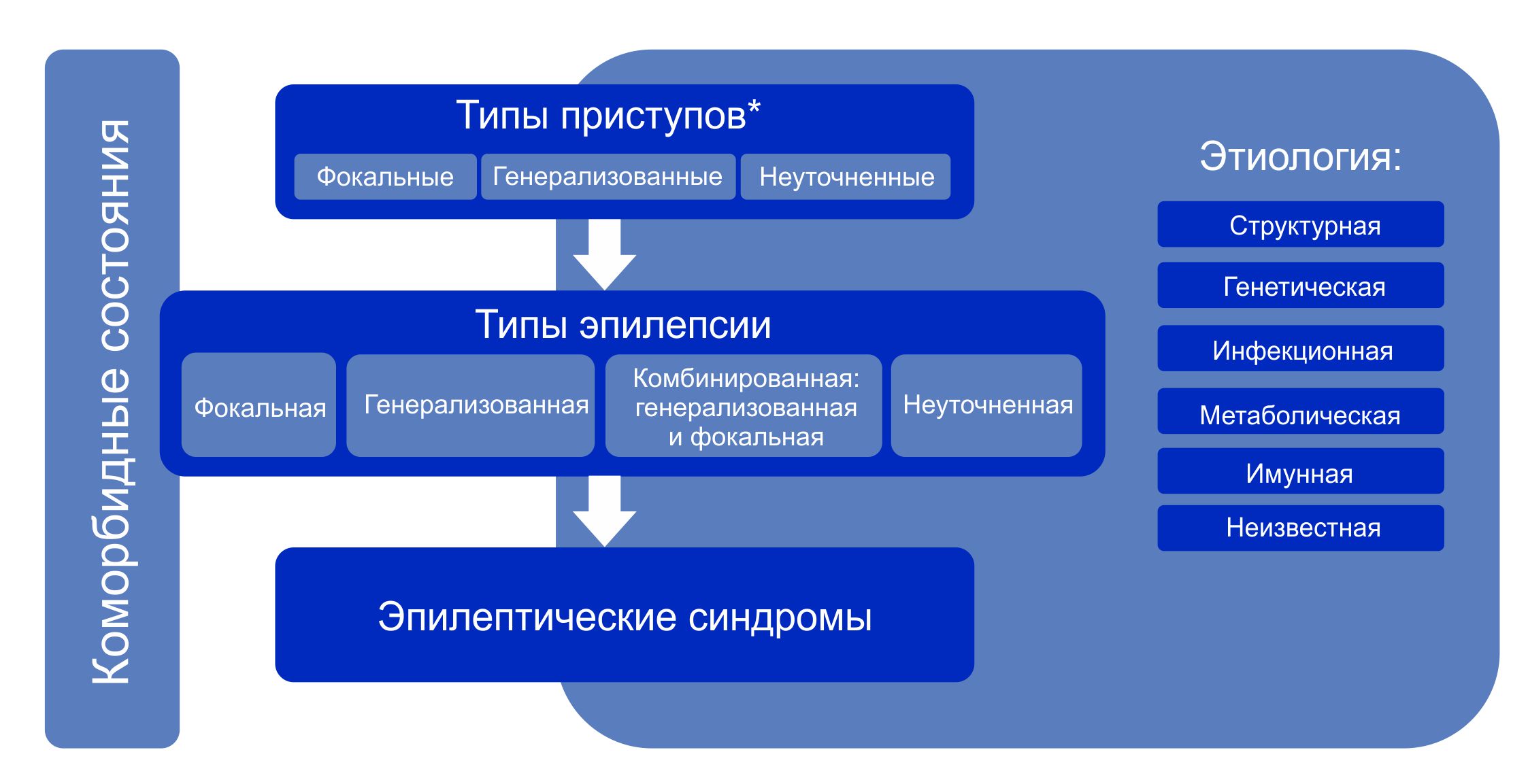
Примечание. * Оценивается по началу приступа.
Авакян Г. Н. Блинов Д. В. Лебедева А. В. Бурд С. Г. Авакян Г. Г. Классификация эпилепсии Международной Противоэпилептической Лиги: пересмотр и обновление 2017 года. Эпилепсия и пароксизмальные состояния. 2017; 9 (1): 6–25. DOI: 10.17749/2077–8333.2017.9. 1.006–025.
Алгоритм классификации эпилепсии:
- На первом этапе (уровне) определяют тип приступа: фокальный, генерализованный или с неизвестным началом.
- На втором этапе (уровне) устанавливают тип эпилепсии: фокальная, генерализованная или сочетанная фокальная и генерализованная, или неизвестная (unknown).
- На третьем этапе (уровне) определяют эпилептический синдром и коморбидность. Эпилептический синдром представляет собой совокупность характеристик, включая тип приступа, данные ЭЭГ и нейровизуализации, часто имеет возрастзависимый характер, провоцирующие факторы, хронозависимость и в ряде случаев определенный прогноз.
- На четвертом этапе (уровне) устанавливают этиологию эпилепсии: структурная, генетическая, инфекционная, метаболическая, иммунная или с неизвестной этиологией.
Пациенты с генерализованной эпилепсией имеют генерализованные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ паттерны, которые сопровождают генерализованные типы приступов (например, генерализованный спайк-волна). Сопутствуют семейная история генерализованных типов приступов или генерализованная эпилепсия.
Пациенты с фокальной эпилепсией имеют фокальные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ-паттерны, которые сопровождают фокальные типы приступов (такие как фокальные острые волны или фокальное интериктальное замедление). Нейроимиджинг демонстрирует фокальную структурную аномалию мозга и способствует установке диагноза, хотя пациенты с генетической этиологией и нормальной визуализацией могут также иметь фокальную эпилепсию. Фокальные эпилепсии могут быть унифокальными, мультифокальными или полушарными.
Пациенты могут иметь как генерализованные, так и фокальные типы приступов, с интериктальными и / или иктальными ЭЭГ-паттернами, которые сопровождают оба типа приступов. Пациенты с синдромом Драве и синдромом Леннокс-Гасто могут иметь генерализованную и фокальную эпилепсию.
В то время как концептуализация эпилепсий по их этиологии очень важна, эпилепсии также могут быть организованы (в соответствии с установленными достоверными общепринятыми клиническими и ЭЭГ — характеристиками) в эпилептические синдромы. Такие синдромы имеют типичный возраст начала приступа, специфические типы приступов и характеристики ЭЭГ и часто другие признаки, которые вместе взятые позволяют диагностировать конкретный эпилептический синдром. Идентификация синдрома эпилепсии полезна, так как она предоставляет информацию о том, какие основные этиологии следует учитывать и какие антиконвульсанты могут быть наиболее полезными. Некоторые эпилептические синдромы демонстрируют аггравацию приступов при определенных антиконвульсантах, чего можно избежать при ранней диагностике синдрома эпилепсии.
Неонатальный и младенческий период:
- самокупирующиеся неонатальные приступы и самокупирующаяся семейная неонатальная эпилепсия;
- самокупирующаяся семейная и несемейная младенческая эпилепсия;
- ранняя миоклоническая энцефалопатия;
- синдром Отахара;
- синдром Веста;
- синдром Драве;
- миоклоническая эпилепсия младенчества, младенческая эпилепсия с мигрирующими фокальными приступами;
- миоклоническая энцефалопатия при непрогрессирующих заболеваниях;
- фебрильные приступы, фебрильные приступы плюс, генетическая эпилепсия с фебрильными приступами плюс.
Детский возраст:
- эпилепсия с миоклонически-атоническими (ранее – астатическими) приступами;
- синдром Леннокса–Гасто;
- фебрильные приступы, фебрильные приступы плюс;
- абсансная эпилепсия;
- эпилепсия с миоклоническими абсансами;
- детская затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса);
- детская затылочная эпилепсия с поздним дебютом (синдром Гасто);
- фотосенситивная затылочная эпилепсия;
- самокупирующаяся эпилепсия с центрально-темпоральными спайками;
- атипичная эпилепсия с центрально-темпоральными спайками;
- эпилептическая энцефалопатия с продолженной пик-волновой активностью во время сна;
- синдром Ландау–Клеффнера;
- аутосомно-доминантная ночная лобная эпилепсия.
Подростковый и взрослый возраст:
- юношеская абсансная эпилепсия;
- юношеская миоклоническая эпилепсия;
- эпилепсия с изолированными генерализованными тонико-клоническими приступами;
- аутосомно-доминантная эпилепсия со слуховыми проявлениями;
- другие семейные височные эпилепсии.
Любой возраст:
- семейная фокальная эпилепсия с вариабельным фокусом;
- рефлекторные эпилепсии;
- прогрессирующие миоклонические эпилепсии.
Наиболее важные генетические причины эпилепсии, которые могут быть идентифицированы при клиническом тестировании:
- хромосомные аномалии;
- аномалии гена.
Существует много способов, которыми генетические факторы могут способствовать развитию эпилепсии. Определенные генетические факторы, возможно, не были унаследованы и не могут быть переданы потомству. Вот некоторые важные генетические концепции, используемые на этом веб-сайте, и их определения:
- унаследованные аномалии генов, аутосомно-доминантное, аутосомно-рецессивное и менделевское наследование;
- приобретенные аномалии генов — de novo, спорадические, мозаицистические, зародышевые и соматические;
- полигенная / комплексная генетическая этиология.
Структурные эпилепсии определяются как имеющие выраженную структурную аномалию мозга, которая связана с существенно повышенным риском эпилепсии. Структурная аномалия мозга может быть приобретена (например, вследствие инсульта, травмы или инфекции) или может быть генетического происхождения; однако, как мы это понимаем в настоящее время, структурная аномалия мозга представляет собой отдельное нарушение, расположенное между приобретенным или генетическим дефектом и эпилепсией.
Магнитно-резонансная томография (МРТ) с использованием 1.5 Тесла аппарата является минимальным стандартным исследованием для исключения структурной аномалии. При этом исследовании важно выполнять протоколы, специфические для эпилепсии, которые позволяют тщательно изучать специфические приобретенные аномалии (например, склероз гиппокампа ) и тонкие пороки развития коры головного мозга, такие как фокальная дисплазия коры. Изображение с использованием 3 Тесла и использование передовых методов анализа программного обеспечения может помочь в выявлении структурных нарушений, не очевидных при обычной МРТ. Интериктальная и иктальная ЭЭГ вместе с дополнительными функциональными исследованиями нейровизуализации, такими как ПЭТ, ОФЭКТ и МЭГ, помогают внимательно изучить конкретную область мозга и идентифицировать тонкую аномалию. У детей младшего возраста в возрасте до 2 лет тонкие аномалии не могут быть выявлены из-за незаконченной миелинизации, и повторное исследование требуется в динамике.
Общие структурные аномалии мозга, связанные с эпилепсией:
- пороки развития коры головного мозга;
- сосудистые пороки развития;
- гиппокампальный склероз;
- гипоксически-ишемические структурные аномалии;
- травматическая повреждение мозга;
- опухоли;
- порэнцефалическая киста.
Метаболические эпилепсии определяются как имеющие определенное метаболическое нарушение, связанное с выраженным риском развития эпилепсии. Метаболические расстройства имеют генетическое происхождение; однако, как мы это понимаем в настоящее время, метаболические аномалии представляют собой отдельное нарушение, стоящее между генетическим дефектом и эпилепсией.
Важные метаболические эпилепсии:
- дефицит биотинидазы и голокарбоксилазы-синтазы;
- дефицит церебрального фолата;
- нарушения креатина;
- приступы при нарушении фолатного цикла;
- недостаточность транспортера глюкозы 1 (GLUT1);
- митохондриальные расстройства;
- пероксисомальные расстройства;
- пиридоксинзависимая эпилепсия.
Иммунные эпилепсии определены как имеющие выраженную иммунную опосредованную этиологию с подтверждением воспаления центральной нервной системы, что, как было показано, связано с существенно повышенным риском развития эпилепсии.
Важные иммуноопосредованные эпилепсии:
- Синдром Расмуссена;
- Антителоопосредованная эпилепсия.
Наиболее распространенная этиология эпилепсии во всем мире является инфекционной, особенно в развивающихся странах. Инфекции в центральной нервной системе могут вызывать как острые симптоматические припадки (которые тесно связаны со сроками первичной инфекции), так и эпилепсией. Инфекционная этиология включает туберкулез, ВИЧ, церебральную малярию, нейроцистицеркоз, подострый склерозирующий панэнцефалит, церебральный токсоплазмоз. Эти инфекции иногда имеют структурный коррелят, однако основная причина эпилепсии определяется как инфекционный процесс. Инфекционная этиология может иметь специфические последствия лечения. Существуют также последствия для общественного здравоохранения, поскольку профилактика таких инфекций может снизить нагрузку на эпилепсию, особенно в развивающихся странах. Наиболее распространенные из таких инфекций следующие:
- бактериальный менингит или менингоэнцефалит;
- церебральная малярия;
- церебральный токсоплазмоз;
- цитомегаловирусная инфекция;
- ВИЧ;
- нейроцистицеркоз;
- туберкулез;
- вирусный энцефалит;
- подострый склерозирующий панэнцефалит;
- прочие инфекции (токсокариоз, шистосомоз, болезнь Лайма (нейроборрелиоз).
Существует ряд состояний, связанных с рецидивирующими пароксизмальными событиями, которые могут имитировать симптомы, и ошибочно диагностироваться как эпилепсия. Важно, чтобы эти расстройства учитывались при оценке пароксизмальных событий, так как частота ошибочных диагнозов при эпилепсии высока во всем мире. История заболевания и видеозапись приступов помогают установить правильный диагноз. Существуют некоторые состояния, при которых могут сосуществовать эпилептические и неэпилептические события.
- вазовагальный обморок;
- рефлекторные аноксические приступы;
- аффективно-респираторные апноэ;
- гипервентиляционный обморок;
- самоиндуцированный обморок по методу Вальсальвы;
- неврологический обморок (мальформация Киари, гиперэксплексия, пароксизмальное болевое нарушение);
- насильственная верхняя непроходимость дыхательных путей;
- ортостатическая интолерантность;
- удлинение QT и сердечный обморок;
- одышечно-цианотические обмороки (при тетраде Фалло).
- мечтательность / невнимательность;
- самоудовлетворение;
- эйдетические образы;
- вспышки и реакции ярости;
- ощущения вне тела;
- панические атаки;
- диссоциативные состояния;
- неэпилептические приступы;
- галлюцинации при психических расстройствах;
- выдуманная/поддельная болезнь.
- связанные со сном ритмические двигательные нарушения;
- гипнотические вздрагивания;
- парасомнии;
- нарушения сна в фазу REM-сна;
- доброкачественный неонатальныймиоклонус сна;
- периодические движения ног;
- нарколепсия-катаплексия.
- тики;
- стереотипии;
- пароксизмальная кинезиогенная дискинезия;
- пароксизмальная некинезиогенная дискинезия;
- пароксизмальная дискинезия, вызванная нагрузкой;
- окулогирный криз;
- эпизодические атаксии;
- альтернирующая гемиплегия;
- гиперэксплексия;
- синдром опсоклонус-миоклонуса.
- мигрень со зрительной аурой;
- семейная гемиплегическая мигрень;
- доброкачественный пароксизмальный тортиколлис;
- доброкачественное пароксизмальное головокружение;
- циклическая рвота.
- доброкачественный миоклонус младенчества и дрожательные атаки;
- синдром Сандифера;
- неэпилептические падения головы;
- spasmus nutans;
- gовышенное внутричерепное давление;
- cемейный синдром ректальной боли;
- cпинальный миоклонус.
С современных позиций определяется 5 этапов:
- описание пароксизмального события, возможно по данным анамнеза;
- классификация приступа (анамнез, клиника, ЭЭГ, видео-ЭЭГ мониторинг);
- диагностика формы эпилепсии (анамнез, клиника, ЭЭГ, видео-ЭЭГ мониторинг, нейровизуализация);
- установление этиологии эпилепсии (МРТ, кариотипирование, биохимические исследования, биопсия мышц и прочее);
- диагностика сопутствующих заболеваний и установление инвалидности.
Читайте также:
