
Эпилептическая энцефалопатия с продолженной спайк волновой активностью во сне











Гормональная терапия эпилептических синдромов с продолженной спайк-волновой активностью во сне
В настоящий момент гормональная терапия эпилептических синдромов с продолженной спайк-волновой активностью во сне является перспективным развивающимся направлением эпилептологии. Отдельные клинические примеры и глобальные мета-анализы неизменно подтверждают эффективность гормонов в тех случаях, когда не удается достигнуть успеха с помощью антиэпилептических препаратов. Однако до сих пор не разработаны единые подходы к выбору препарата и схемы его назначения. В данной статье рассмотрены возможные механизмы действия стероидов. Приведены показания к применению гормонального лечения, сведения об эффективности и переносимости. На основе собственного опыта предложен протокол дифференцированной тактики гормонального лечения эпилептических синдромов с продолженной спайк-волновой активностью во сне. Выбранная тактика позволяет добиваться положительного эффекта в отношении снижения числа эпилептических приступов и предотвращать развитие тяжелого нейрокогнитивного дефицита у 70–80% пациентов.
Энцефалопатия с электрическим эпилептическим статусом в фазу медленного сна, или ESES (сокр. от англ. Encephalopathy with electrical status epilepticus during slow sleep), – состояние, при котором не столько эпилептические приступы (ЭП), а сама эпилептиформная (межприступная) активность способствует нарушению высших психических функций. Данное состояние характеризуется неоднородностью клинических проявлений и специфической электроэнцефалографической картиной в виде продолженной спайк-волновой активности в фазу медленного сна (сокр. CSWS от англ. Сontinuous spike and waves during sleep) [1]. В настоящее время границы двух определений ESES и CSWS стерты, они часто употребляются как синонимы [1–3]. ESES/CSWS – широкое понятие, объединяющее спектр близких по клинической картине эпилептических синдромов, к которым относятся непосредственно эпилептическая энцефалопатия с продолженной спайк-волновой активностью, синдром Ландау–Клеффнера (ЛКС) и атипичная эволюция доброкачественных фокальных эпилепсий детства (роландической и редко – синдрома Панайотопулоса).
При данной патологии основным неблагоприятным фактором прогноза являются не ЭП, которых в отдельных случаях может и не быть, а приобретенные интеллектуальные и двигательные нарушения, которые становятся необратимыми при продолжительности заболевания более 18 месяцев [2].
Цель лечения синдромов с ESES/CSWS – скорейшее подавление эпилептической активности и, соответственно, ЭП, что предупреждает дальнейшую нервно-психическую деградацию. Тем не менее, назначая лечение, следует понимать, что при чрезмерно длительном существовании ESES/CSWS улучшение на электроэнцефалограмме (ЭЭГ) уже не обеспечивает нормализацию психоречевого развития. В этом случае целью терапии будет уменьшение числа ЭП и/или предупреждение дальнейшего регресса. Вернуть нормальное психоречевое развитие ребенку в случае позднего начала адекватной терапии уже не удается.
Наиболее наглядным доказательством эффективности ГТ являются данные ретроспективного мета-анализа лечения 575 пациентов с ЕSES/CSWS, проведенного в 2015 г. (рис. 1) [3]. Исследование подтвердило, что наиболее эффективный подход – это применение именно ГТ, которая оказалась эффективнее антиэпилептических препаратов (АЭП), в т.ч. бензодиазепинов. В резистентных к ГТ случаях прибегают к хирургическому лечению, которое позволяет улучшать состояние большинства пациентов.

Согласно данным литературы, показаниями к назначению ГТ при синдромах с ЕSES/CSWS являются [2, 3]:
- неэффективность или недостаточная эффективность АЭП;
- частые ЭП и/или статусное течение приступов;
- нейрокогнитивный регресс, а именно появление двигательных, поведенческих или речевых нарушений (даже в отсутствие ЭП).
Наиболее перспективным направлением в изучении точки приложения стероидов является разработка модели воспаления. В 2011 г. группой ученых были проведены исследования, доказавшие повреждение гематоэнцефалического барьера при эпилепсии [4]. При магнитно-резонансной томографии головного мозга было показано появление очагов изменения сигнала, напоминающих таковые при воспалении и соответствующих локализации эпиактивности на ЭЭГ. При изучении эпилептического статуса у крыс, предварительно получивших дексаметазон, количество особей с развившимся эпилептическим статусом сократилось, а время до его начала значительно увеличилось по сравнению с группой контроля, а также снизилась смертность экспериментальных животных. Результаты исследования позволили сформулировать гипотезу о том, что восстановление целостности гематоэнцефалического барьера может быть одним из механизмов, участвующих в противоэпилептическом действии глюкокортикостероидов (ГКС) [4].
Другая группа авторов в своих исследованиях сосредоточилась на эффектах нейростероидов [5]. Было высказано предположение, будто если эндогенные гормоны при катамениальной эпилепсии могут влиять на частоту возникновения судорог, то экзогенные гормоны также могут быть способными изменять возбудимость нейронов. В экспериментальных моделях эпилепсии были показаны возбуждающие нейрональные эффекты эстрогена и ингибирующее действие прогестерона [6]. Важно отметить, что влияние прогестерона на фармакоиндуцированные ЭП было дифференцированным в зависимости от пола: у самок прогестерон имел противосудорожные эффекты, в то время как у самцов эффект был проконвульсантным. Это можно объяснить влиянием многих факторов, в т.ч. наличием полового диморфизма в областях мозга, ответственных за генерацию и контроль ЭП, и в медиаторных системах [6].
По мнению других ученых, эффект применения ГТ может быть обусловлен увеличением периода полураспада АЭП, повышением их концентрации в крови, усилением постсинаптического действия, ингибированием высвобождения возбуждающих аминокислот и поддержанием стабильного мембранного потенциала [7].
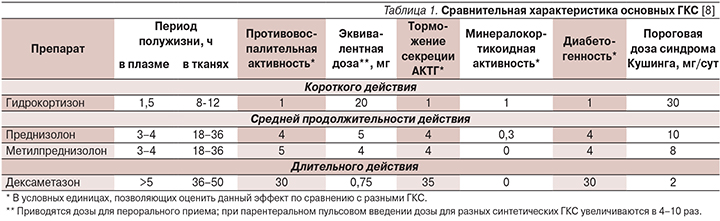
В лечении синдромов с продолженной спайк-волновой активностью применяют как гормоны коры надпочечников с преимущественно глюкокортикоидной функцией – гидрокортизон, так и их синтетические аналоги – преднизолон, метилпреднизолон, дексаметазон и в редких случаях – тетракозактид (синтетический адренокортикотропный гормон). Ниже приведена табл. 1, содержащая данные, позволяющие дифференцированно подходить к выбору препарата и его доз.
При разных ежедневных схемах приема ГКС следует назначать первую дозу в ранние утренние часы (между 6 и 8 часами); если же одноразовый прием невозможен из-за величины дозы, 2/3 дозы назначаются в 8 часов и 1/3 – днем (около полудня) [8].
Эффективность ГТ оценивается по уменьшению числа ЭП (или их полному прекращению) и по позитивной динамике на ЭЭГ – уменьшение спайк-волнового индекса во сне, а также по позитивной динамике психических и речевых функций и поведения ребенка.
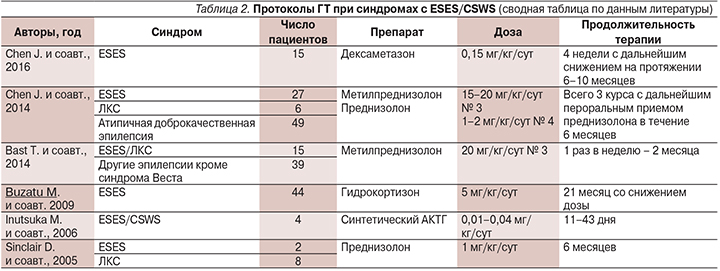
Среди применяемых препаратов лучше всего изучен метилпреднизолон. Последним и самым крупным является ретроспективное исследование J. Chen и соавт. (2014), включившее 82 пациента (табл. 2). Все они получили три курса метилпреднизолона в виде внутривенных инфузий в течение трех дней, с последующим пероральным приемом преднизолона в течение четырех дней. После трех курсов пероральный прием преднизолона был продолжен до 6 месяцев. Время персистирования CSWS cоставило от 2 до 10 лет. В результате общая эффективность достигла 83% с максимумом среди пациентов с синдромом Ландау–Клеффнера [9]. Эффективность метилпреднизолона была доказана и по результатам другого ретроспективного исследования, где предлагалась иная схема ГТ, причем не только для пациентов с ESES (табл. 2) [11]. Она включала четыре курса метилпреднизолона в дозе 20 мг/кг/сут по 3 дня каждую неделю. После четырех недель у 30 из 54 (56%) пациентов в общей выборке и у 11 из 15 (73%) – с ESES и синдромом Ландау–Клеффнера получен положительный эффект. Средняя продолжительность терапии составила 11 недель.
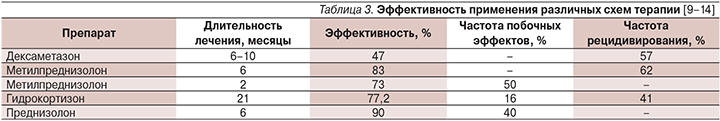
Данные по протоколам (выбор препарата, его доза и продолжительность лечения) остальных исследований по ГТ синдромов с ESES/CSWS также приведены в табл. 2. В табл. 3 представлены показатели эффективности лечения, частоты рецидивирования и развития побочных эффектов гормональных препаратов в зависимости от длительности курса.
Из данных, приведенных в табл. 3, следует, что гидрокортизон не уступает по эффективности метилпреднизолону и его применение вполне целесообразно [12]. Улучшение было достигнуто 34 из 44 (77,2%) пациентов, включая нормализацию ЭЭГ у 21 пациента, получавших лечение гидрокортизоном по 5 мг/кг/сут с постепенным снижением дозы на протяжении 21 месяца [12]. В то же время дексаметазон может по своей эффективности уступать метилпреднизолону.
Для оценки эффективности применения ГТ было проведено собственное ретроспективное исследование, включившее 43 пациента в возрасте от 2 до 14 лет с синдромами с ESES/CSWS (индекс продолженной спайк-волновой активности во сне составил 50% и более). Метилпреднизолон назначался сначала в виде пульсовой терапии: внутривенно капельно в дозах 25–30 мг/кг/сут в течение 3 дней, затем по 15 мг/кг/сут в течение 2 дней. Продолжительность пульсовой терапии в 5 дней обусловлена тем фактом, что именно в эти сроки (от третьего до пятого дня), как правило, происходит сокращение числа или прекращение ЭП. В те же сроки наблюдается сокращение частоты эпилептиформных разрядов, которое можно увидеть и на обычной рутинной 20-минутной ЭЭГ бодрствования (записывать ЭЭГ сна в такие короткие сроки в обычной клинической практике не всегда представляется возможным в силу трудоемкости исследования).
После первого курса пульс-терапии пациенты были разделены на две группы: в первую вошли дети без значительных нейрокогнитивных нарушений (n=17), во вторую – больные с выраженными нейрокогнитивными и/или речевыми нарушениями и тяжелым течением заболевания (n=26). Пациентам первой группы были проведены повторные курсы пульс-терапии 1 раз в месяц в аналогичных дозах на протяжении еще 5 месяцев. Пациентам второй группы после окончания первого курса инфузионной терапии был назначен пероральный прием метилпреднизолона по 1,5 мг/кг/сут с постепенным снижением дозы на протяжении 5 месяцев. Таким образом, общая продолжительность ГТ в обеих группах составила 6 месяцев. Положительная динамика оценивалась по сокращению числа ЭП более чем на 50% или их полному купированию, снижению индекса эпилептической активности, улучшению нейрокогнитивных функций и речи. Эффективность применения метилпреднизолона составила 76 и 80% в первой и второй группах соответственно, а в целом при эпилептических синдромах с ESES/CSWS – 79% (34/43).
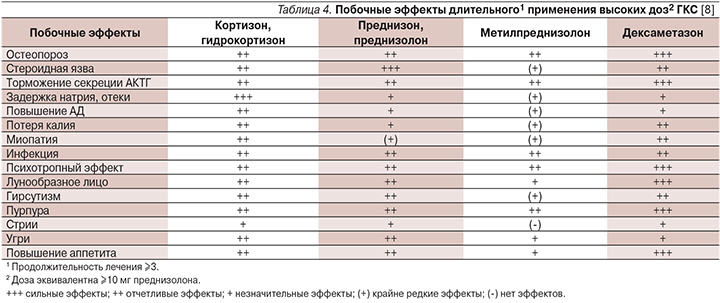
В табл. 4 представлены данные о возможных побочных эффектах ГТ и времени их появления. Родители детей, получающих ГТ, должны быть информированы о мерах профилактики побочных эффектов: соблюдение гипокалорийной рациональной диеты, прием препаратов калия, контроль уровня глюкозы в крови и артериального давления, раннее назначение антибактериальной терапии при инфекционных заболеваниях. Также необходимы контроль минерального обмена (биохимическое исследование крови) и ранняя профилактика остеопороза.
Жизнеугрожающих осложнений не отмечено ни в одном из исследований, представленных в табл. 4, все негативные реакции имели временный характер и полностью регрессировали по завершении лечения.

По мнению большинства авторов, самый короткий курс ГТ не должен быть менее 3 месяцев, в среднем он занимает от 6 месяцев до 1,5 лет [1–3]. После 3 месяцев приема гормонов снова проводится оценка эффективности и при необходимости – коррекция терапии. Собственный опыт и литературные данные свидетельствуют о том, что неэффективность одного типа гормонального препарата не является предиктором резистентности к ГТ в целом. Смена препарата может позволить достигнуть положительного эффекта. Это наглядно показано на примере терапии дексаметазоном [10]. Из 15 пациентов с ESES/CSWS от 7 был получен эффект после 4-недельного лечения дексаметазоном. Важно отметить, что из 7 больных, ответивших на терапию, 5 ранее были резистентными к терапии преднизолоном в сравнимых дозах. Можно предположить, что это явление связано с отличиями в химической структуре метаболитов применяемых лекарственных средств и, соответственно, с различными механизмами их действия [10].
Негативной стороной ГТ синдромов с ESES/CSWS является значительная частота рецидивирования заболевания. После прекращения лечения частота рецидивов колеблется в диапазоне от 30 до 78%, и в основном они развиваются в течение 2 месяцев после прекращения терапии [10]. В таких ситуациях рекомендуется прибегать к повторным курсам ГТ. Пока нет данных о том, что повторная ГТ по выбору препарата или дозам должна отличаться от первоначального курса [10–14].
В заключение стоит отметить, что потенциальная польза от назначения ГТ при синдромах с продолженной спайк-волновой активностью во сне превосходит возможные побочные эффекты этого лечения. Она часто позволяет вернуть ребенку нормальную речь и когнитивные функции, а также позволяет контролировать ЭП. В отсутствие адекватного лечения синдромов с ESES/CSWS ЭП становятся реже или прекращаются только к периоду полового созревания, замещение продолженной спайк-волновой активности физиологическими паттернами наступает примерно к 11 годам, но приобретенные нейропсихологические нарушения различной степени выраженности сохраняются на всю жизнь.
- ГТ является наиболее эффективным методом лечения синдромов с продолженной спайк-волновой активностью во сне. Прогноз заболевания тем лучше, чем раньше начато лечение и чем менее выражен нейрокогнитивный дефицит.
- Показаниями к ГТ при синдромах с ESES/CSWS являются неэффективность или недостаточная эффективность АЭП, частые ЭП и/или статусное течение приступов, нейрокогнитивный регресс (появление двигательных, поведенческих или речевых нарушений даже в отсутствие ЭП).
- Соотношение терапевтического потенциала и частоты побочных эффектов позволяет утверждать, что одним из препаратов выбора для длительной терапии синдромов с ESES/CSWS является метилпреднизолон.
- ГТ должна иметь длительный характер (не менее 6 месяцев). В отсутствие положительного эффекта через 3 месяца терапии необходимо прибегнуть к смене препарата (на другой ГКС).
- Строгая профилактика развития побочных эффектов ГТ может позволить минимизировать их частоту.
- После завершения гормонального лечения пациент в течение не менее 6 месяцев должен находиться под наблюдением невролога с регулярным проведением ЭЭГ сна и нейропсихологического тестирования.
1. Berg A.T., Berkovic S.F., Brodie M.J. Buchhalter J., Cross J.H., van Emde Boas W., Engel J., French J., Glauser T.A., Mathern G.W., Moshé S.L., Nordli D., Plouin P., Scheffer I.E. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51(4):676–85.
2. Kramer U., Sagi L., Goldberg-Stern H., Zelnik N., Nissenkorn A., Ben-Zeev B. Clinical spectrum and medical treatment of children with electrical status epilepticus in sleep (ESES). Epilepsia. 2009;50:1517–24.
3. Van den Munckhof B., Van Dee V., Sagi L., Caraballo R.H., Veggiotti P., Liukkonen E., Loddenkemper T., Sánchez Fernández I., Buzatu M., Bulteau C., Braun K.P., Jansen F.E. Treatment of electrical status epilepticus in sleep: A pooled analysis of 575 cases. Epilepsia. 2015;56(11):1738–46.
4. Marchi N., Granata T., Freri E., Ciusani E., Ragona F., Puvenna V., Teng Q., Alexopolous A., Janigro D. Efficacy of anti-inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistantepileptics. PLoS One. 2011;28;6(3):e18200.
5. Velíšková J., Desantis K.A. Sex and hormonal influences on seizures and epilepsy. Horm. Behav. 2013;63(2):267–77.
6. Stevens S.J., Harden C.L. Hormonal Therapy for Epilepsy. Curr. Neurol. Neurosci. Rep. 2011;11:435–42.
7. Yuan Q., Li F., Zhong H. Early diagnosis, treatment and prognosis of epilepsy with continuous spikes and waves during slow sleep. Int. J. Clin. Exp. Med. 2015;8(3):4052–58.
9. Chen J., Yang Z., Liu X., Ji T., Fu N., Wu Y., Xiong H., Wang S., Chang X., Zhang Y., BaoX., JiangY., Qin J. Efficacy of methylprednisolone therapy for electrical status epilepticus during sleep in children. Zhonghua Er Ke Za Zhi. 2014;52(9):678–82.
10. Chen J., Cai F., Jiang L., Hu Y., Feng C. A prospective study of dexamethasone therapy in refractory epileptic encephalopathy with continuous spike-and-wave during sleep. Epilepsy Behav. 2016;55:1–5.
11. Bast T, Richter S., Ebinger F., Rating D., Wiemer-Kruel A., Schubert-Bast S. Efficacy and tolerability of methylprednisolone pulse therapy in childhood epilepsies other than infantile spasms. Neuropediatrics. 2014;45(6):378–85.
12. Buzatu M., Bulteau C., Altuzarra C., Dulac O., Van Bogaert P. Corticosteroids as treatment of epileptic syndromes with continuous spike-waves during slow-wave sleep. Epilepsia. 2009;50(7):68–72.
13. Inutsuka M., Kobayashi K., Oka M., Hattori J., Ohtsuka Y. Treatment of pilepsy with electrical status epilepticus during slow sleep and its related disorders. Brain Dev. 2006;28(5): 281–86.
Медицинский консультативно-диагностический центр по вопросам диагностики и лечения эпилепсии и других пароксизмальных состояний Альфа-ритм
+7 (343) 287-55-05, +7-912-655-31-90
Детские эпилептические энцефалопатии
Перунова Н.Ю., невролог-эпилептолог, д.м.н.
Выраженность резидуального дефицита, формирующегося в исходе этих заболеваний, пропорциональна продолжительности активного периода. Критической длительностью активного периода эпилептических энцефалопатий с CSWS (ЭЭ с CSWS) предложено считать срок от 2 до 3 лет, после чего полноценное восстановление интеллектуальных функций невозможно.
В качестве отрицательных факторов прогноза ЭЭ с CSWS выделяют ранний возраст дебюта (до 3 лет), большую продолжительность активной фазы течения заболевания (более 3 лет), низкий уровень и длительное время диагностики, отсутствие ответа на терапию. Прогноз нарушений речи при эпилептических энцефалопатиях с CSWS считается неблагоприятным при дебюте заболевания в возрасте до 3 лет, сохранении речевых нарушений более 1 года.
В течении ЭЭ с CSWS предложено выделять активный период, характеризующийся персистированием паттерна CSWS, и резидуальный период. Проспективные исследования, содержащие информацию о динамике течения ЭЭ с CSWS, немногочисленны. Анализируются данные по группам из 11-18 пациентов (детей, подростков, молодых взрослых), длительность наблюдения от 5 до 9 лет, максимально – 16 лет.
Основными синдромами ЭЭ с CSWS являются наличие эпилептических припадков, расстройства поведения, когнитивные нарушения, нарушения речи, изменения на ЭЭГ.
Эпилептические припадки возникают у 80-90% пациентов, страдающих ЭЭ с CSWS. Назначение антиконвульсантов оказывается эффективным в отношении контроля припадков в 60% случаев, однако воздействие на эпилептиформную активность является недостаточным В отдаленном периоде эпилептические припадки могут сохраняться у 14% пациентов.

Рис.1. Сравнительная частота различных видов эпилептических припадков у пациентов с ЭЭ с CSWS (Перунова Н.Ю., Томенко Т.Р., 2004). ПП – простые парциальные; ВГСП – вторично-генерализованные судорожные припадки; АА – атипичные абсансы; ПАД. – эпилептические падения; СП – сложные парциальные припадки.


Расстройства поведения в острой фазе ЭЭ с CSWS наблюдаются у 90% детей, однако при проспективном наблюдении у 75% из них отмечается постепенное снижение выраженности поведенческих нарушений.
Когнитивные нарушения в активном периоде ЭЭ с CSWS встречаются у 90% пациентов. Хорошее восстановление когнитивных функций возможно только у 10%, обучение в массовой школе – у 12%. Выраженный интеллектуальный дефицит в отдаленном периоде сохраняется у 18% подростков и молодых взрослых.
Полный регресс нарушений речи в исходе ЭЭ с CSWS возможен в 17-18% случаев, частичное восстановление нарушений речи — в 37-40%. У 50-60% пациентов речевые нарушения сохраняются в течение всей жизни.
Продолженная эпилептиформная активность с индексом более 50% — облигатный симптом ЭЭ с CSWS. В активном периоде эпилептиформная активность оценивается как диффузная в 40%, случаев, как диффузная с региональными преобладанием — в 60%. Клинические проявления ЭЭ с CSWS и характеристики эпилептиформной активности не всегда совпадают Персистирование изменений на ЭЭГ при LKS регистрируется до 5 лет, при ESES до 9 лет.


Рис.5. Динамика основных синдромов ЭЭ с CSWS (собственная реконструкция по данным метаанализа).
К сожалению, доступные в настоящее время терапевтические воздействия на ЭЭ с CSWS недостаточно эффективны, что заставляет считать проблему ЭЭ с CSWS одной из наиболее актуальных и нерешенных в детской эпилептологии.
(Обзор составлен по данным публикаций: Smith MC. et al., 2003; Rossi PG. et al., 2004; Nickels K, Wirrell E., 2006; Tassinari CA, Rubboli G., 2006; Van Hirtum-Das M. et al., 2006; Wang S. et al., 2006; Praline J, et al., 2006; Scholtes FB. et al., 2010; García-Peñas JJ., 2010; Liukkonen E. et al., 2010; Loddenkemper T. et al., 2011.)

Cиндромы c CSWS являются функциональными расстройствами, возникающими во время активного синаптогенеза с аксональным спраутингом. Нейрональная активность или установление синаптических контактов существенно влияет на то, какие из этих синапсов останутся, а какие редуцируются до достижения 10-летнего возраста. Постоянная эпилептиформная активность во время медленного сна в период организации мозга негативно влияет на установление соответствующих нейрональных связей, нормальное его развитие и функционирование.
Сноска 1. Постоянная эпилептиформная активность во время медленного сна в период организации мозга негативно влияет на установление соответствующих нейрональных связей, нормальное его развитие и функционирование.
Эпилептические энцефалопатии, связанные с CSWS
Эпилепсия с электрическим эпилептическим статусом во время медленного сна
Согласно недавнему предложению ILAE [7] данный синдром ESES определяется как частично обратимая, возраст-зависимая детская эпилептическая энцефалопатия, характеризующаяся следующими признаками:
1) фокальные и генерализованные эпилептические приступы (унилатеральные или билатеральные клонические, тонико-клонические, абсансы, парциальные моторные, сложные парциальные приступы и эпилептические падения);
2) нейропсихологические нарушения в форме общей или избирательной регрессии когнитивных функций (исключением является приобретенная афазия, которая традиционно рассматривается отдельно);
3) двигательная недостаточность в форме атаксии, нарушении целенаправленных движений, дистонии или односторонних поражений;
4) ЭЭГ паттерны диффузных спайк-волн (унилатеральных или фокальных) занимающие не менее 85% медленного сна и персистирующие в трех записях ЭЭГ в течение, по крайней мере 1 месяца.
Необходимым условием для диагностики этого синдрома является наличие постоянных спайк-волн во время медленного сна.
Демографические данные. Синдром ESES наблюдается только у детей. Частота его составляет 0,5% от общего числа детей с эпилепсией, с преобладанием у мальчиков (63%).
Сноска 2. Синдром ESES встречается только в детском возрасте с частотой 0,5% от общего числа детей с эпилепсией с преобладанием у мальчиков (63%).
Этиология. Этиология неизвестна. Более чем у одной трети пациентов причиной ECSWS могут быть врожденные мальформации и перинатальное повреждение головного мозга [8]. При проведении нейровизуализации патологию головного мозга можно обнаружить в 60% случаев.
Сноска 3. При проведении нейровизуализации у пациентов с ECSWS патология головного мозга выявляется в 60% случаев.
Стадия относительной нормализации ЭЭГ
Через несколько лет после достижения ремиссии эпилепсии отмечается постепенное улучшение ЭЭГ. Разряды во время медленного сна становятся более короткими, менее частыми и более фрагментарными. Различимыми становятся физиологические паттерны сна. Однако на ЭЭГ сна могут длительно сохраняться редкие фокальные комплексы острых и медленных волн, даже после клинического улучшения. Нормализация ЭЭГ, если она достигается, может занимать более 15 лет [6].
Диагностика. Проведение МРТ головного мозга обязательно. Более чем у трети пациентов выявляются изменения на МРТ. Наиболее типичными являются кортикальная атрофия, порэнцефалия, нарушения кортикального развития.
Функциональная визуализация головного мозга (PET или SPECT) обычно выявляет патологию даже у пациентов с нормальной МРТ.
Электроэнцефалографическое исследование включает в себя проведение стандартной ЭЭГ, продолженного видео-ЭЭГ мониторинга или амбулаторного мониторинга. Подозрение на заболевание может возникнуть при записи ЭЭГ во время короткого сна, но для более точного определения необходима регистрация ЭЭГ на протяжении всего сна.
Дифференциальная диагностика. Дифференциальная диагностика ЕCSWS проводится с синдромом Ландау-Клеффнера [13]. При синдроме Ландау-Клеффнера доминирующим когнитивным нарушением является приобретенная афазия, эпилептические приступы могут отсутствовать, и на интериктальной ЭЭГ фокусы эпилептиформной активности локализуются преимущественно в височных отведениях, в то время как при ECSWS – преимущественно в лобных.
Следует проводить дифференциальный диагноз с синдромом Леннокса-Гасто, отличительной особенностью которого являются тонические приступы с быстрой пароксизмальной активностью на ЭЭГ, которые при ECSWS отсутствуют. Моторные приступы и ремиссии при синдроме Леннокса-Гасто крайне редки.
Прогноз. Спонтанное разрешение эпилептиформных разрядов на ЭЭГ и эпилептических приступов происходит в подростковом возрасте, что совпадает со стабилизацией или улучшением нейропсихологических и поведенческих функций [6]. Но выздоровление всегда происходит медленно и, в большинстве случаев, лишь частично. Персистирование и тяжесть резидуальных поведенческих, когнитивных и речевых нарушений зависит от возраста начала и продолжительности активной фазы эпилептиформной активности.
Приобретенная эпилептическая афазия (синдром Ландау-Клеффнера) – детское заболевание, характеризующееся ассоциацией приобретенной афазии и мультифокальных спайков и спайк-волновых разрядов на ЭЭГ.
Демографические данные. Возраст дебюта заболевания составляет 2-8 лет, с пиком в 5-7 лет. Мальчики болеют в 2 раза чаще. В специализированные центры поступают один или два новых пациента в год.
Этиология неизвестна. Наследственность отягощена по эпилептическим приступам в 12% случаев и в 5% в тех случаях, если пациенты не имеют приступов. Однако есть сообщения о найденных на биопсии аномалиях головного мозга у 3% пациентов с синдромом Ландау-Клеффнера [8].
Клиническая манифестация. Первым симптомом обычно является слуховая вербальная агнозия. Дети перестают идентифицировать звуки окружающей среды и соотносить их с предметами, что делает их похожими на больных с аутизмом. Родители замечают, что дети не реагируют даже на громкое обращение. Слуховая вербальная агнозия может прогрессировать и осложняться импрессивной и экспрессивной афазией с полной утратой произносимой речи. Диагностика, как правило, ошибочна, т.к. первоначально ставится диагноз приобретенной глухоты или элективного мутизма. Многим детям записывают аудиограмму, которая оказывается нормальной. Лингвистический дефицит может быть выявлен не сразу из-за выраженного нарушения поведения и когнитивных проблем.
Таким образом, главным когнитивным нарушением, описываемым как приобретенная афазия, является аудиторная вербальная агнозия, возникающая у изначально здоровых детей, навыки психоречевого развития и словарный запас которых соответствуют возрасту.
Начало заболевания подострое. Отмечается постепенное ухудшение речевых функций в виде изменения экспрессивной речи, появления парафазий, стереотипий, персевераций и фонологических ошибок. Ребенок начинает выражаться телеграфным стилем или очень простыми предложениями, в конечном итоге становится полностью немым и перестает реагировать на невербальные сигналы.
Один из необъяснимых признаков синдрома Ландау-Клеффнера – это флюктуация симптоматики речевых нарушений, характеризующаяся ремиссиями и ухудшениями.
Познавательные и поведенческие расстройства наблюдаются у 75% пациентов. Наиболее характерными являются дефицит внимания с гиперактивностью. В редких случаях это может прогрессировать до тяжелой расторможенности и психоза. Длительное исследование этих пациентов в динамике показало, что интеллектуальная недостаточность развивается не во всех случаях.
Эпилептические приступы наблюдаются также у 75% пациентов, но они редкие и имеют хороший прогноз. Дебют приступов наблюдается в возрасте 4-6 лет. Только у 20% пациентов приступы продолжаются после 10 лет, но до 15 лет полностью исчезают.
Приступы полиморфные: фокальные моторные, атипичные абсансы, атонические приступы с падениями, малые автоматизмы и вторично-генерализованные. Кратковременные приступы с минимальными моторными и субъективными симптомами могут быть частыми, но они трудноуловимы и могут не определяться. В 1/3 случаев отмечаются только генерализованные тонико-клонические приступы или изолированный эпилептический статус, который может возникать в возрасте 5-10 лет. Сложные парциальные приступы, характерные для лобнодолевого региона, исключены, так же как и тонические приступы.
Приступы чаще ночные, редкие, хорошо поддаются лечению и полностью исчезают в возрасте 13-15 лет.
Диагностика. МРТ головного мозга, как правило, не выявляет никаких отклонений. Но функциональная нейровизуализация показывает изменения в височной доле [14]. При проведении объемного МРТ анализа выявляется уменьшение объема верхней височной извилины [15], в которой локализована речевая зона.
ЭЭГ характеризуются в основном фокусами комплексов острая-медленная волна в задневисочных отведениях, которые часто могут быть мультифокальными и билатерально синхронизированными, существенно усиливающимися при NREM сне. CSWS возникает на определенной стадии заболевания почти во всех случаях, но это не является главным требованием при постановке диагноза, т.к. может сохраняться или исчезать в течение REM сна.
Дифференциальная диагностика. Многие случаи синдрома Ландау-Клеффнера ошибочно принимают за глухоту, аутизм или другие психиатрические заболевания.
Возникновение острой или подострой прогрессирующей афазии у детей 2-8 лет без приобретенного пареза или симптомов энцефалита наиболее вероятно связано с синдромом Ландау-Клеффнера.
Синдром Ландау-Клеффнера также трудно отличить от ECSWS из-за схожих клинических признаков и данных ЭЭГ. Однако главным отличием являются выраженные речевые нарушения при синдроме Ландау-Клеффнера, в то время как при ECSWS главным критерием для постановки диагноза является наличие CSWS на ЭЭГ [16].
Прогноз. Эпилептические приступы, изменения на ЭЭГ исчезают к 15 годам. К этому возрасту также значительно уменьшаются речевые и нейропсихологические расстройства и половина всех больных могут вести относительно нормальную жизнь. Около 10-20% пациентов полностью выздоравливают.
Существует прямая связь между продолжительностью CSWS и степенью нарушения речевых функций [17]. Ранний дебют заболевания связан с плохим прогнозом для речевых функций.
Сноска 4. Ранний дебют синдрома Ландау-Клеффнера коррелирует с плохим прогнозом для речевых функций в дальнейшем.
Очень редко встречаются спонтанные ремиссии через несколько недель или месяцев после начала заболевания.
Лечение синдромов с ESES
Эпилептические приступы при ECSWS и синдроме Ландау-Клеффнера не частые, ограничены возрастом и легко контролируются назначением антиконвульсантов. Однако медикаментозное лечение направлено в первую очередь на уменьшение эпилептиформной активности на ЭЭГ, что уменьшает речевые, поведенческие и когнитивные расстройства.
Единого протокола лечения синдромов с CSWS не разработано. Все традиционные антиконвульсанты не дают удовлетворительного результата. Препараты, предназначенные для лечения фокальных эпилепсий (фенитоин, карбамазепин, вигабатрин) противопоказаны, т.к. могут вызвать значительное усиление эпилептиформной активности и, соответственно, усугубить нейропсихологический дефицит.
Лечение синдромов с CSWS начинается с подбора противоэпилептической терапии. Используются антиконвульсанты широкого спектра действия (вальпроаты: вальпроат натрия (депакин), вальпроевая кислота (конвулекс), леветирацетам (кеппра)) как в монотерапии, так и в сочетании с этосуксимидом (суксилепом), клоназепамом или клобазамом (фризиумом) [5,12,18]. Сультиам (осполот) является препаратом выбора. Клобазам и сультиам на сегодняшний день в России не зарегистрированы.
Были предложены следующие схемы лечения: пероральный прием бензодиазепинов (диазепама, клобазама, клоназепама) в сочетании с вальпроатом короткими циклами (3-4 нед.) [5,12,18]. При применении ректального диазепама (1 мг/кг) наблюдается улучшение [18].
Если лечение антиконвульсантами не дает эффекта, рекомендован АКТГ (тетракозактид (синактен-депо) по 2 мл ежедневно или с интервалами до 3 дней и постепенным уменьшением дозы в течение 3 мес.) или большие дозы кортикостероидов (преднизолон 2-5 мг/кг ежедневно с постепенным уменьшением дозы в течение 3 мес.) [5], особенно у новых пациентов [12]. Продолжительность лечения после этого периода зависит от результатов и побочных эффектов. АКТГ и стероиды применяются вместе с вальпроатами и бензодиазепинами, прием которых продолжается после отмены гормональной терапии.
В некоторых случаях успешно применяется внутривенный иммуноглобулин.
Эффективность лечения оценивается как клинически, так и при помощи повторной регистрации ЭЭГ сна. В случае рецидива CSWS после ранее успешно проведенного курса гормональной терапии целесообразно его повторить.
В резистентных случаях синдрома Ландау-Клеффнера успешно применяется субпиальная интракраниальная транссекция. Эта хирургическая техника позволяет убирать способность кортикальной ткани распространять эпилептиформную активность и генерировать разряды, сохраняя нормальные кортикальные физиологические функции.
Источник: журнал "Медицинский совет" №3-4 2008.
Читайте также:
