
Генерализованные миоклонические эпилептические приступы это

Миоклоническая форма эпилепсии характеризуется припадками, при которых не возникает физических болевых ощущений.
Такое заболевание в основном затрагивает детей, хотя в редких случаях и в определенных видах проявляется у взрослых 20-30 лет.
Болезнь чаще носит врожденный характер, но может развиваться у здоровых детей под действием определенных внешних и внутренних факторов.
Описание
Заболевание в МКБ-10 обозначается кодом G40.4.
Патология в основном проявляется в возрасте 13-18 лет и характеризуется миоклоническими приступами, которые выражаются в непроизвольных подергиваниях или активности, приступы при этом всегда затрагивают парные органы в верхней части тела.

Реже судороги наблюдаются в нижних конечностях.
Основная форма болезни – миоклонус-эпилепсия, которая представляет собой наследственное поражение центральной нервной системы, за счет чего и случаются миоклонические припадки.
Также известна форма юношеской миоклонической эпилепсии, которая называется синдром Янца.
Это генерализованная доброкачественная форма патологии, которая возникает на фоне возрастных трансформаций в организме.
Наиболее подвержены риску развития такого недуга лица в возрасте от 8 до 25 лет, но чаще всего в группу риска входят дети 12-15 лет.
Все проявления миоклонической эпилепсии в зависимости от происхождения можно условно разделить на три группы:
- симптоматическая. Заболевание является следствием других патологических процессов в организме;
- идиопатическая. Патология передается наследственным путем и проявляется в детском или подростковом возрасте;
- криптогенная. Точно определить этиологию таких заболеваний не удается. Такие формы эпилепсии наиболее тяжело поддаются медикаментозному лечению.
Некоторые криптогенные формы со временем переходят в одну из двух других групп, так как с развитием уровня диагностической медицины иногда становится возможным определение причины развития болезни.
Формы и стадии миоклонуса
Заболевание классифицируется в зависимости от степени тяжести и сопутствующей симптоматики и может относиться к одному из следующих видов:

Происхождение болезни не всегда удается определить. Основным признаком ДМЭМ являются припадкообразные сокращения мышц шеи, конечностей и головы.
Такие припадки случаются у детей в возрасте от полугода до трех лет два-три раза в день и длятся не более трех секунд, поэтому обратить внимание на наличие заболевания не всегда удается своевременно.
Данная форма легче всего поддается лечению, но при полном отсутствии возможно проявление осложнений в виде отставания в развитии психомоторных функций.
Заболевание поражает одного из 40 000 младенцев и считается очень редкой формой и также в основном имеет неопределенную этиологию.
Принято считать, что патология проявляется на фоне воспалительных процессов в ЦНС, что приводит к нарушению проводимости мозговых импульсов, отсюда и эпилептические припадки.
Признаки заболевания становятся очевидны в возрасте до одного года.

Преимущественно наследственная форма, которая проявляется в быстро прогрессирующих и учащающихся эпилептических припадках (в основном они развиваются при воздействии внешних раздражителей).
Болезнь тяжело поддается лечению и в основном применяется симптоматическая терапия, которая при удачном стечении обстоятельств позволяет почти полностью замедлить развитие патологии, но окончательное ее устранение невозможно.
Симптоматика недуга проявляется в период от 5 до 16 лет или позже, и сначала эпилептические припадки как таковые отсутствуют: у ребенка наблюдается только подергивание мимических лицевых мышц, что часто ошибочно воспринимается как нервный тик.
С возрастом подергивания переходят на мышцы плечевого пояса и на верхние конечности, и спровоцированы такие приступы могут быть такими внешними факторами, как резкие вспышки света, стрессовые ситуации и резкие чрезмерные физические нагрузки.
При отсутствии лечения с годами развиваются такие осложнения, как нарушения речи, поражения мышц лица, атаксия и тремор.
Заболевание также обозначается аббревиатурой MERRF (myoclonic epilepsy, ragged-red fibres).
Заболевание возникает как следствие мутаций в транспортной РНК лизина, передающихся по материнской линии.
При таком нарушении транспортная РНК передает внутри клеток неточную информацию, что приводит к нарушению клеточной деятельности, а она в свою очередь является причиной такого неврологического поражения, как эпилепсия.

Заболевание MERRF является единственным в группе, которое может проявляться не только у детей, но и у взрослых людей до самой старости.
Патология быстро прогрессирует и проявляется в виде эпилептических припадков и судорог, деменции, ухудшению с годами слуха и зрения, но своевременное симптоматическое лечение дает положительные результаты в большинстве случаев.
Причины возникновения у взрослых и детей
Сегодня чаще говорят о наследственных причинах развития миоклонической эпилепсии, особенно если речь идет о младенческих формах.
Удалось выявить определенные гены, в которых локализуются патогенные участки, при передаче которых и развивается заболевание, но это далеко не все возможные фрагменты ДНК, при мутациях в которых может появиться такая болезнь.
В целом недуг всегда обусловлен генетическими нарушениями, но передаваться они могут не только наследственным путем, но и на фоне следующих болезней и провоцирующих факторов:
- воздействие на плод во время беременности токсических веществ, попадающих в организм матери при курении, употреблении алкоголя и наркотических или лекарственных препаратов;
- внутриутробное инфицирование (особенно вирусами, которые способны легко нарушать структуру генов);
- воздействие радиации на плод.
Патология может развиваться в эмбрионе и при сахарном диабете или патологиях легких и сердца, которые у будущей матери протекают в хронической форме. В таких случаях часто происходят сбои в формировании центральной нервной системы плода.
Три этапа течения

Вне зависимости от формы, заболевание развивается в три этапа.
Первый – эпилептически-титаниформный – проходит в течение нескольких лет, на протяжении которых развиваются и прогрессируют эпилептические припадки.
Дополнительными симптомами являются нарушения психики, при которых пациент часто испытывает беспричинный страх, приступы могут сопровождаться болезненными спазмами, у человека повышается потливость. Припадки всегда проходят без потери сознания.
Второй период – миоклонический-эпилептический. Его развитие тоже занимает годы, а приступы хотя и не оказывают существенного негативного влияния на жизнедеятельность, распространяются не только на мышцы лица, но и на другие части тела, поражая целые группы мышечных волокон.
В тяжелой стадии развития этого этапа человек утрачивает способность обслуживать себя самостоятельно, нарушается ориентация в пространстве. Приступы эпилепсии могут быть спровоцированы любым резким звуком или вспышкой света.
Таким людям противопоказано употребляь алкоголь, так как приступы могут возникать под действием спиртного. В целом они могут появиться в любое время даже при эмоциональном напряжении, и единственное время, когда приступы исключены – это ночной сон.
Последняя, терминальная стадия, отличается усиливающимися описанными симптомами, но при этом у пациента происходят серьезные нарушения психики.
Обычно когнитивные функции не утрачиваются полностью, но у человека явно заметны расстройства памяти и речи, и иногда таких людей можно принять за слабоумных.
Симптоматика заболевания

При такой форме эпилепсии неспециалисту достаточно сложно определить, что у пациента начался припадок, особенно на начальных стадиях.
Но если припадки случаются – им предшествует непроизвольное подергивание верхних конечностей и сокращение мышц лица, шеи и головы, при этом пациент находится в сознании.
Наиболее вероятное время развития припадков – раннее утро, сразу после пробуждения.
Припадки могут случаться раз в день или раз в несколько месяцев, интенсивность таких приступов может быть разной – от подергиваний века глаза до сильных судорог верхних конечностей.
Типы припадков
Припадки при данном заболевании развиваются не всегда, но если они случаются – их всегда можно классифицировать следующим образом:
- Миоклонические. Проявляются в виде подергиваний рук, мышц плечевого пояса и шеи, подергивания глаз и век. В течение дня (за исключением утренних часов) такие припадки могут не проявляться, но к вечеру в случае сильной усталости и переутомления они возникают.
- Тонико-клонические. Такие приступы внешне выглядят так же, как миоклонические, но отличаются по природе происхождения: они возникают при нарушениях мозговой деятельности, связанных с неправильным режимом сна.
- Абсансы. Вид припадков, при котором пациент теряет сознание на несколько секунд, и в течение этого времени исчезают и судорожные проявления.
От них требуется выполнить все предписания, касающейся первой помощи эпилептикам, и вызвать врача.
Диагностика

Лечение эпилепсии и ее диагностика – сложные процессы, которые требуют участия невролога и психиатра.
Но при подозрении на миоклоническую форму в первую очередь необходимо выполнить полное обследование, чтобы становить тип и степень тяжести патологи и разработать корректный терапевтический курс.
Для этого в обязательном порядке выполняются следующие процедуры:
- Проводится сбор анамнеза: опрашивается сам пациент и его близкие родственники с целью выявления случаев неврологических расстройств в роду (для определения возможной передачи патологии генетическим путем).
- Выполняются МРТ и КТ головного мозга. Выполняются для обнаружения и оценки нарушений структур головного мозга.
- Замеряется уровень электрической активности головного мозга с помощью электроэнцефалографии.
На основании полученных результатов назначаются соответствующие терапевтические процедуры.
Схема лечения
Эффективность миоклинической эпилепсии в настоящее время низкая, и даже в случае успешного подбора медикаментозных препаратов лучшим исходом будет купирование симптомов, но не полное устранение проблемы.
Медикаментозное лечение является основной, но не единственной формой лечения. В данном случае наиболее эффективно применение противосудорожных препаратов:
- Клоназепам;
- Фенобарбитал;
- Конвулекс;
- Бензонал;
- Депакин.
Обычно такое лечение длится как минимум два года, и учитывая, что все используемые препараты обладают показателем токсичности как минимум выше среднего, пациентов и их родственников заранее предупреждают о возможных осложнения и побочных эффектах.

В связи с такими рисками врачи никогда не ориентируются исключительно на медикаментозное лечение, и помимо устранения судорог проводится семейная и социальная терапия, корректируется режим бодрствования и сна.
Часто пациенту требуется устранение физиологических причин развития припадков (в том числе – удаление образований в области головного мозга: примерно в 10% случаев болезнь возникает на фоне появления таких опухолей).
Первая помощь при приступе
На поздних стадиях такого заболевания у пациента могут случаться серьезные припадки и приступы, в том числе – и в общественных местах, при этом человек может оказаться не в состоянии помочь себе самостоятельно.
Если свидетелем эпилептических приступов становятся посторонние – с их стороны необходимы следующие действия:
О миоклонической эпилепсии в этом видео:
Осложнения, прогнозы и профилактика
Прогнозы лечения заболевания – умеренно-благоприятные, если терапию начать своевременно. В таких случаях возможно практически полное устранение симптоматики и припадков до уровня локальных нервных тиков.
Формально можно сказать, что в этом случае эпилепсия вылечена, но если терапия проводится с запозданием или назначаются неправильные препараты – средняя продолжительность жизни пациента составляет около 20 лет (хотя некоторые доживают до глубокой старости даже с сильными припадками).
К летальным исходам в таких случаях приводят осложнения эпилепсии: это пневмония, кахексия и другие присоединенные патологии.

Младенческие формы патологии чреваты нарушения психики и отставанием детей в интеллектуальном развитии.
Особых действенных профилактических рекомендаций, которые позволили бы избежать развития болезни, не существует, но снизить риск этого можно, если в детском возрасте ограничивать потребление кофеиносодержащих продуктов и употреблять в пищу больше фруктов и овощей, которые богаты растительной клетчаткой.
Миоклонические припадки иногда свойственны здоровым людям. Часто они наблюдаются при серьезных физических нагрузках (нередко это можно заметить у спортсменов), но если такое происходит у взрослых – в основном причин для беспокойства нет.
Но если такие признаки, как подергивание глаз, мышц лица и рук наблюдаются в детском возрасте и без очевидных предпосылок – следует показать ребенка врачу.
Возможно, это позволит вовремя распознать болезнь и начать лечение, благодаря которому патологические осложнения будут ведены к минимуму.
Об ошибках в лечении миоклонической эпилепсии в этом видео:
Эпилептические припадки встречаются у людей различного возраста. Генерализованная судорожная эпилепсия характеризуется возникновением характерных эпиприступов – абсансов, либо генерализованных миоклонических и тонико-клонических пароксизмов. Для выявления заболевания рекомендуется проводить электроэнцефалографию, компьютерную или магнитно-резонансную томографию. Лечение генерализованной формы патологии основывается на использовании монотерапии или комбинированного лечения с использованием антиконвульсантов.
Причины возникновения
Основная причина идиопатической генерализованной эпилепсии – наличие генетических дефектов. Наиболее часто, наследственные изменения затрагивают гены, ответственные за формирование каналов на мембранах нервных клеток. Это приводит к нестабильности нейронов и их легком возбуждении на любые, даже обычные стимулы. Считается, что при наличии болезни у одного из родителей, риск рождения ребенка с генерализованной эпилепсией достигает 5-10%. Выделяют два варианта заболевания, в зависимости от числа “поврежденных” генов – моногенный и полигенный.
Симптоматический вариант патологии характеризуется появлением эпилептических припадков на фоне органического поражения головного мозга. В этом случае развитие эпиприступов может быть связано с черепно-мозговыми травмами, энцефалитом, менингитом, доброкачественными и злокачественными опухолями головного мозга, приобретенными и врожденными нарушениями метаболизма. Кроме того, в детском возрасте эпилепсия возникает после родовых травм, длительных гипоксических состояний и наличии аномалий развития коры больших полушарий. Следует отметить, что симптоматическая эпилепсия чаще носит фокальный вариант течения, в то время как генерализованные приступы характерны для идиопатической эпилепсии.
Внимание! Прогноз для больного зависит от варианта патологии, а также времени начала лечения.
Отличия типов генерализованной эпилепсии
Идиопатическая, или криптогенная генерализованная эпилепсия
Впервые возникает у детей и подростков. Первичные приступы чаще всего выявляются в возрасте до 20-22 лет. Кроме эпиприпадков первично-генерализованного характера клинические проявления отсутствуют. При тщательном неврологическом осмотре специалист может выявить диффузную симптоматику. Очаговые симптомы не характерны. Личность и когнитивные функции ребенка полностью сохранены, так как органические дефекты в головном мозге отсутствуют. Современные исследования показывают, что у небольшого числа детей (около 5%) возможны легкие интеллектуальные нарушения.
Симптоматическая эпилепсия
Может возникнуть в любом возрасте, так как заболевания головного мозга, обусловленные внешними причинами, не имеют какого-либо возрастного деления. Если же болезнь связана с врожденными аномалиями структур ЦНС или метаболическими заболеваниями, то вторичная форма может манифестировать у детей. Клинические проявления включают в себя не только генерализованные приступы, но также очаговую и общемозговую симптоматику, связанную с первичной патологией (ЧМТ, нейроинфекции и др.). При возникновении симптоматической эпилепсии в детском возрасте характерна олигофрения.
Варианты клинических проявлений
В клинической практике принято выделять три формы генерализованных пароксизмов: типичный абсанс, генерализованные тонико-клонические приступы и генерализованные миоклонические приступы. Они имеют различные внешние проявления, что помогает проводить дифференциальную диагностику.
Типичный абсанс
Характеризуется краткосрочной утратой сознания. Продолжительность периода – от 1 до 40-50 секунд. Больной внешне замирает и прекращает выполнять какую-либо деятельность. При наличии вегетативного компонента возможно покраснение или побледнение лица, повышенное слюнотечение. Абсансы характерны для детей. Во время сложного приступа появляются бессознательные движения: облизывание губ, сокращение отдельных мимических мышц, застегивание и расстегивание пуговиц и пр. Родители могут считать, что их дети просто задумались о чем-то в процессе игры, так как клонические или тонико-клонические генерализованные сокращения мышц отсутствуют.
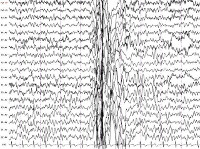
При типичном абсансе наблюдаются специфические изменения на электроэнцефалограмме (ЭЭГ) – появление генерализованных пик-волновых комплексов со средней частотой 3 Гц. Частота во время припадка постепенно снижается. Если абсанс носит атипичный характер, т. е. сопровождается моторным компонентом, то у пик-волновых комплексов отмечают нерегулярную частоту.
Генерализованная эпилепсия с тонико-клоническими приступами
Характеризуется двухфазностью проявлений. Сначала развивается тоническая фаза, проявляющаяся тоническим напряжением мускулатуры, с последующим переходом в клоническую фазу. Последняя связана с прерывистыми мышечными сокращениями. Весь эпиприступ протекает на фоне потери сознания. Приступ первично-генерализованной эпилепсии начинается с внезапного падения пациента, после которого сразу же наблюдаются тонические сокращения. Продолжительность первой фазы – до 40 секунд. Клоническая фаза длится до 4-6 минут. Эпиприступ заканчивается непроизвольным мочеиспусканием и резким падением мышечного тонуса. Больной как правило засыпает, однако, сон достаточно короткий – до 1 часа. Во время сна возможны тонические или клонические пароксизмы отдельных мышц.
Генерализованные миоклонические приступы
В случае генерализованных миоклонических приступов возникают не ритмичные мышечные подергивания, развивающиеся в результате хаотичного сокращения мышечных волокон в различных группах мышц. Важная черта пароксизма – подергивания имеют симметричный характер, однако, могут не затрагивать все тело, в отличие от генерализованного тонико-клонического пароксизма. Человек во время эпиприступа сохраняет сознание, однако плохо реагирует на окружающие события, окружающих и их речь. При проведении ЭЭГ отмечается возникновение симметричных множественных пик-волновых комплексов с различной частотой.
Диагностические мероприятия

Для выявления первично генерализованной эпилепсии врачи проводят комплексную диагностику, основанную на сборе клинических данных и результатов ЭЭГ. На энцефалограмме больных с идиопатическим вариантом патологии изменения в базовом ритме не характерны, однако волны мозговой активности могут быть замедлены. В отличие от этого, при вторично генерализованной эпилепсии основной ритм всегда нарушен, что связано с постоянным присутствием патологического очага (опухоли, кисты и др.). Во время приступа в обоих случаях выявляются характерные изменения в мозговой активности, связанные с пик-волновыми комплексами, имеющими симметричный и синхронный характер.
При подозрении на вторичный характер болезни, например у больных с редкими генерализованными припадками, проводится компьютерная или магнитно-резонансная томография головного мозга. Данные методы позволяют оценить структуры ЦНС и выявить в них патологические очаги (рост новообразований, формирование кист, очаги инфильтрациями клеток при нейроинфекциях). Консультация генетика показана всем больным с первичной формой. При возможности проводится исследование генов, связанных с эпилепсией, с помощью ПЦР-диагностики или методов секвенирования. Диагноз “идиопатическая эпилепсия” выставляет после того, как все возможные органические причины развития будут исключены.
При диагностике генерализованной эпилепсии следует проводить ее дифференциальную диагностику с:
- обмороками, связанными с заболеваниями внутренних органов или вегетативной нервной системы;
- гипогликемией;
- пароксизмами, обусловленными истерическим неврозом или шизофренией;
- сомнамбулизмом и др.
Вопросами диагностики и интерпретацией полученных результатов занимается только врач-невролог. Самостоятельно выставлять диагноз и подбирать лечение не следует.
Эффективное лечение
Основная цель терапии идиопатической эпилепсии – добиться полного исчезновения эпиприступов или существенно снизить их количество. Качество жизни пациента напрямую зависит от частоты эпилептических пароксизмов. Кроме того, эффективная антиэпилептическая терапия определяет дальнейший прогноз.
В медицинской практике выделяют несколько групп антиконвульсантов, которые имеют различный механизм действия, что влияет на их эффективность и возможные противопоказания. Наиболее часто используют следующие медикаменты:
- Блокаторы натриевых каналов, к которым относят Ламотриджин, Трилептан и Карбамазепин. Ламотриджин уменьшает количество глутамата в нервных синапсах, тем самым снижая возбудимость нейронов и частоту пароксизмов. В отличие от Ламотриджина, Карбамазепин напрямую блокирует натриевый канал и нарушает процесс передачи возбуждения от нейрона к нейрону.
- Препараты, увеличивающие чувствительность нервных клеток к ГАМК: Диазепам и Фенобарбитал. ГАМК – тормозной медиатор в головном мозге. При повышении чувствительности нейронов к данному веществу повышает порог возбудимости, что снижает бесконтрольное возникновение возбуждения.
- Ингибиторы кальциевых каналов (Этосуксимид) блокирует распространение нервного возбуждения по отросткам нервных клеток, препятствуя формированию эпилептических очагов в коре головного мозга.
- Модуляторы синаптических везикул 2А-протеина – современная группа медикаментозных средств, оказывающих комплексное действие на нейроны и снижающих их возбудимость. Одобренным для клинической практики считается препарат Леветирацетам.
- Вальпроевая кислота и ее производные – классические антиконвульсанты, однако, механизм их действия не до конца понятен. По-видимому, данные медикаменты комплексно блокируют рецепторы на поверхности нервной клетки.

Наиболее распространенная форма применения лекарственных препаратов для лечения генерализованной эпилепсии – таблетки и капсулы. Это позволяет эффективно продолжать лечение в домашних условиях, так как проведение инъекций не требуется. На фармацевтическом рынке существуют медикаменты с замедленным высвобождением действующего вещества, что позволяет снизить кратность их применения и повысить безопасность при длительном использовании.
Внимание! Если больной использует любые другие лекарственные средства для лечения сопутствующих заболеваний, необходимо предупредить об этом лечащего врача.
Терапия генерализованной эпилепсии проводится по специальному алгоритму, обеспечивающему повышение эффективности и снижение рисков развития побочных эффектов:
- Монотерапия – основной подход для идиопатической формы патологии. При первично выявленном заболевании терапия всегда должна назначаться с использования одного препарата. Лечение начинают с минимальной терапевтической дозировки. Если пароксизмы сохраняются, дозу увеличивают, обязательно сохраняя ее в безопасных пределах. Неэффективность лечения на фоне увеличения дозировки является показанием к смене медикаментозного средства. К комбинированному использованию антиконвульсантов прибегают после двух неудачных попыток монотерапии. Связано это с тем, что все препараты данной группы имеют выраженные побочные эффекты. При одновременном назначении 2 и более лекарств, риск развития нежелательных последствий существенно возрастает.
- Всем больным показано титрование дозы. Данное понятие связано с тем, что начальная дозировка всегда в несколько раз ниже средних значений, используемых у людей с эпилепсией. Чувствительность пациентов к антиконвульсантам индивидуальна и варьируется в значительных пределах.
- Выбор препарата при первой манифестации эпилептических пароксизмов зависит от возраста пациента и наличия сопутствующих патологий. Врач может выбирать медикамент с любым механизмом действия. Дополнительный ограничивающий фактор – стоимость препаратов и финансовые возможности человека.
- Лечение генерализованной эпилепсии продолжается от 2 лет до пожизненного использования антиконвульсантов. Для оценки эффективности используют дневник больного, в котором он должен фиксировать пароксизмы, а также контрольное проведение электроэнцефалографии. При правильном подборе антиконвульсанта и титровании дозы, исчезновение эпиприступов наблюдается у 75% больных.
- Врач определяет сроки отмены терапии. Ни в коем случае не следует самостоятельно менять дозировку лекарственных средств или полностью отказываться от них. Это может стать причиной прогрессирования патологии и увеличения частоты возникновения пароксизмов.
Помимо лекарственной терапии всем пациентам рекомендуется посещать психотерапевта, нормализовать образ жизни, режим труда и отдыха. Важно отметить, что при вторичном характере эпилепсии необходимо устранить ее непосредственную причину появления. Если заболевания развилось на фоне опухоли головного мозга, то проводят ее хирургическое удаление, лучевую терапию или используют химиотерапевтические средства.
Прогноз
Прогноз генерализованной эпилепсии зависит от варианта ее развития. В связи с тем, что идиопатическая форма не характеризуется органическим изменением в структурах ЦНС, умственной отсталостью и неврологическими проявлениями, прогноз благоприятный при соблюдении алгоритма лечения. Исходы вторичной формы определяются течением и терапией первичного заболевания.

Миоклоническая эпилепсия — заболевание, основу которого оставляют миоклонические эпилептические пароксизмы. Эпизоды миоклонических судорог у больных сочетаются с генерализованными клонико-тоническими эпиприступами, абсансами. Сопутствующая неврологическая симптоматика зависит от формы эпилепсии. Диагностика включает сбор анамнеза, оценку неврологического и психического статуса, электроэнцефалографию, генеалогический анализ, биохимические исследования, нейровизуализацию. Лечение проводится антиконвульсантами, при резистентности — комбинацией противоэпилептических препаратов.
- Причины миоклонической эпилепсии
- Патогенез
- Классификация
- Симптомы миоклонической эпилепсии
- Осложнения
- Диагностика
- Лечение миоклонической эпилепсии
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Причины миоклонической эпилепсии
Обычно ведущим является генетический фактор. Чёткое аутосомно-доминантное наследование прослеживается при синдроме Драве, аутосомно-рецессивное — в отдельных случаях ранней миоклонической энцефалопатии. Некоторые заболевания имеют полигенное наследование. Локализация генетических дефектов установлена не для всех наследственных форм, исследования в этом направлении продолжаются. К генетически детерминированным патологиям относится и симптоматическая МЭ, возникающая вследствие дисметаболических процессов, обусловленных наличием дефектных генов. Образованию спонтанных мутаций в геноме способствуют:
- Внутриутробные инфекции. Инфекционный процесс неблагоприятно отражается на развитии плода. Особенно опасны вирусные инфекции, поскольку вирусы способны провоцировать аномальную перестройку отдельных генов.
- Хронические заболевания беременной. Сахарный диабет, сердечная недостаточность, хронические заболевания лёгких, эндокринная патология матери приводят к гипоксии, метаболическим расстройствам на ранних стадиях развития зародыша. В результате происходят сбои формирования ЦНС, отдельных механизмов обмена веществ.
- Повышенный радиоактивный фон. Радиация оказывает мутагенное влияние на живые организмы. Развивающийся плод наиболее подвержен подобному воздействию. Следствием является возникновение структурных, дисметаболических, функциональных аномалий, влекущих за собой повышенную эпилептическую активность.
- Прием тератогенных медикаментов. Самолечение, незнание о своей беременности в раннем периоде, медицинская необходимость фармакотерапии приводят к приёму опасных для плода медикаментов. Химические вещества оказывают повреждающее воздействие на отдельные гены, вносят изменения в существующие метаболические механизмы.
- Токсические воздействия на плод.Алкоголизм, наркомания, курение женщины в период беременности сопровождаются проникновением токсических веществ в организм плода. Подобно тератогенным фармпрепаратам они способны повредить отдельный локус генома, в результате возникает миоклоническая эпилепсия.
Патогенез
Идиопатические варианты МЭ развиваются вследствие генетически обусловленной повышенной возбудимости церебральных нейронов, приводящей к эпилептогенной активности мозга. Симптоматическая миоклоническая эпилепсия формируется в результате обменных нарушений, накопления в нервных клетках патологических соединений (полисахаридных включений, прионных белков).
При болезни Лафоры, миоклонической энцефалопатии младенцев повышенная эпиактивность обусловлена дисметаболизмом нейронов в условиях разрастания глиальных элементов (при гибели нейронов, нарушении апоптоза астроцитов). Нейрональная гипервозбудимость вызывает возникновение патологической нервной импульсации, идущей к мышечным волокнам. Результатом являются отдельные мышечные сокращения (миоклонии), тонические, клонические судороги. Различная локализация миоклоний отражает локальное возбуждение разных зон мозговой коры. При диффузном распространении гипервозбуждения возникает клонико-тонический пароксизм с тотальным вовлечением мышечных групп.
Классификация
В основе группировки отдельных видов МЭ лежит этиологический принцип. Согласно Международной классификации эпилепсии 1989 года выделяют 3 основные группы:
- Идиопатические — наследственно обусловленные формы. Характерна манифестация симптоматики в детском/подростковом возрасте. Идиопатическими являются доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ), юношеская миоклоническая эпилепсия (ЮМЭ), болезнь Унферрихта-Лундборга, синдром Драве.
- Криптогенные — не имеющие установленной этиологии. Отличаются выраженной резистентностью к фармакотерапии, наличием сопутствующей очаговой симптоматики, интеллектуального дефицита. К криптогенным относятся эпилепсия с миоклонически-астатическими приступами, эпилепсия с миоклоническими абсансами.
- Симптоматические — возникающие на фоне происходящих в организме патологических процессов. В большинстве случаев обусловлены метаболическими нарушениями. Симптоматическими считаются ранняя миоклоническая энцефалопатия, болезнь Лафоры, миоклонические пароксизмы при подостром склерозирующем панэнцефалите, болезни Крейтцфельдта-Якоба.
Впоследствии были выявлены генетические аспекты возникновения криптогенных форм МЭ. Учитывая результаты исследований, Международное общество неврологов предложило относить ранее считавшиеся криптогенными виды МЭ к идиопатическим.
Симптомы миоклонической эпилепсии
Базовым симптомом выступают пароксизмы миоклоний, затрагивающие различные мышечные группы конечностей, реже — лица, еще реже — туловища. Миоклонии выглядят как мышечные подёргивания, при вовлечении мышц одной группы сокращения приводят к непроизвольным двигательным актам, напоминающим гиперкинезы. Миоклонический эпилептический пароксизм происходит при сохранённом сознании, может протекать с перемещением сокращений по различным мышцам. Миоклоническая эпилепсия характеризуется комбинацией миоклоний с клонико-тоническими приступами и/или абсансами. В зависимости от нозологической формы наблюдаются задержка психического развития, атаксия, пирамидная недостаточность, расстройства мышечного тонуса, зрительные нарушения.
ДМЭМ дебютирует в возрастном периоде от 6 месяцев до 3 лет. Приступы захватывают верхние конечности, лицо, шею, могут имитировать наклон головы, моргание, кивки головой. Заболевание редко сопровождается интеллектуальным снижением. Миоклоническая эпилепсия юношеского возраста (манифестация в возрасте 12-18 лет) отличается присоединением тонико-клонических эпизодов, отсутствием неврологического дефицита. Синдром Драве клинически проявляется на первом году жизни, сопровождается олигофренией, нарушениями поведения, пирамидным дефицитом. Семейная миоклония Унферрихта-Лундборга начинается в 5-16 лет, сочетается с тремором, атаксией, дизартрией, психическими расстройствами.
Миоклоническая эпилепсия симптоматического характера отличается прогрессированием симптоматики, выраженным когнитивным дефицитом, прочими неврологическими нарушениями, наличием проявлений основного заболевания, клинико-лабораторных признаков метаболических расстройств.
Осложнения
Клонико-тонические, астатические приступы осложняются травмированием пациента вследствие падения. Генерализованные судороги с утратой сознания опасны западением языка, перекрытием дыхательных путей и асфиксией. Аспирация слюны, рвотных масс приводит к последующему развитию пневмонии. Длительный миоклонический пароксизм, непрерывно следующие кластерные сокращения перерастают в миоклонический эпистатус. В эпилептическом статусе возможны серьёзные дыхательные расстройства, остановка сердца, развитие отёка головного мозга.
Диагностика
- Сбор анамнестических данных. Большое значение имеет возраст дебюта, характер начала, порядок развития симптоматики.
- Неврологический осмотр. Проводится неврологом, направлен на выявление миоклонических сокращений, очагового дефицита, определение уровня психического развития, степени когнитивных расстройств, оценку психического статуса.
- Электроэнцефалография. У большинства пациентов регистрируются диффузные интериктальные симметричные эпилептогенные разряды, иктальные высокоамплитудные спайки. В ряде случаев для выявления эпиактивности требуется суточный ЭЭГ-мониторинг, проведение провокационных проб (ЭЭГ при вспышках света, гипервентиляции, резких звуковых сигналах). Результаты исследований оцениваются нейрофизиологом, эпилептологом.
- Нейровизуализация. До закрытия родничков осуществляется путём нейросонографии, у детей старше года — при помощи МРТ головного мозга. Взрослым может проводиться МСКТ. Морфологические изменения церебральных тканей характерны для симптоматических МЭ.
- Лабораторные исследования. Производятся при подозрении на наличие обменных расстройств. Включают биохимический анализ крови и мочи, специфические анализы.
- Консультация генетика. Сбор семейного анамнеза, составление генеалогического древа позволяют определить наследственный характер эпилепсии, установить тип наследования.
Дифференциальная диагностика осуществляется с неэпилептическим миоклонусом, отличительной особенностью которого выступает фокальный характер миоклоний, отсутствие реакции на провокацию, нормальная ЭЭГ-картина. Дифференцировка МЭ необходима также с судорожным синдромом инфекционной этиологии, фебрильными судорогами, синдромом Леннокса-Гасто, мозжечковой миоклонической диссинергией Ханта.
Лечение миоклонической эпилепсии
Терапия базируется на антиконвульсантах. Подбор фармпрепарата и дозировки осуществляется индивидуально. Препаратами выбора выступают производные вальпроевой кислоты, обладающие противоэпилептическим эффектом в равной степени в отношении миоклонических, клонико-тонических, абсансных пароксизмов. В фармакорезистентных случаях показано комбинированное лечение вальпроатами, бензодиазепинами, этосуксимидом, барбитуратами, антиконвульсантами нового поколения (топираматом, леветирацетамом). Важным моментом является исключение провоцирующих приступы факторов: резких звуков, вспышек света, эмоциональных всплесков, физических перегрузок, перегреваний.
Прогноз и профилактика
Наиболее прогностически неблагоприятна ранняя миоклоническая энцефалопатия, смертность составляет половину случаев заболевания, остальные дети являются глубокими инвалидами. Миоклоническая эпилепсия при болезни Лафоры, Крейтцфельдта-Якоба плохо поддаётся противоэпилептической терапии, сопровождается прогрессирующим интеллектуальным распадом. ДМЭМ и ЮМЭ отличаются доброкачественным течением, редко приводят к когнитивному дефициту. Более 50% случаев ДМЭ заканчиваются спонтанным выздоровлением.
МЭ не имеет специфических мер профилактики. К мероприятиям, способным предупредить рождение больного ребёнка, относятся планирование беременности, ранняя постановка на учёт, исключение неблагоприятных воздействий на плод. Ведение беременности должно включать разъяснительные беседы с женщиной по поводу необходимости охранительного режима, тератогенной опасности лекарственных средств, пагубного воздействия на будущего ребёнка вредных привычек.
Читайте также:
