
Идиопатическая фотосенситивная затылочная эпилепсия
Распространенность
Низкая (0,4% от всех эпилепсий).
Возраст дебюта заболевания
От 15 месяцев до 19 лет, средний возраст
Пол
Незначительно преобладают мужчины (60%).
Этиология
Является частью доброкачественного синдрома предрасположенности к приступам у детей.
Клинические проявления
Рефлекторные приступы, идентичны спонтанным приступам затылочной эпилепсии: элементарные зрительные галлюцинации, нечеткость зрения или слепота, по отдельности или в сочетании. Реже встречаются девиация глаз и головы, трепетание век, боль в орбитах. Затылочные приступы могут сопровождаться вегетативными симптомами, как при синдроме Панайотопулоса. В редких случаях приступы завершаются вторично-генерализованными тонико-клонические судорогами. Могут иметь место также спонтанные зрительные или другие приступы (миоклонии, абсансы . ГТКП). Есть сообщения о зрительных приступах с необычной иктальной симптоматикой, которая может походить на истерические припадки, мигрень. К постиктальным симптомам относятся головная боль, тошнота и рвота.
Провоцирующие факторы
Мерцающий свет, паттерн-стимуляция. Иногда световые стимулы могут вызывать приступы только в сочетании с другими факторами (волнение, усталость, депривация сна). Частой причиной возникновения приступов являются просмотр мультфильмов и видеоигры.
Интериктальная ЭЭГ
Ритмическая фотостимуляции (РФС) вызывает (1) фотопароксизмальные ответы (ФПО) в виде затылочных спайков/полиспайков, или (2) генерализованные ФПО с затылочным акцентом. Могут регистрироваться также и спонтанные центротемпоральные и затылочные спайки. Зрительные вызванные потенциалы имеют аномально высокую амплитуду.
Иктальная ЭЭГ
Затылочные разряды быстрых спайков, которые или внезапно прекращаются, или распространяются в височные отделы, или переходят в ГТКП.
Дифференциальная диагностика
Мигрень, идиопатическая затылочная детская эпилепсии по типу Гасто (значение провоцирующих факторов), идиопатическая фотосенситивная генерализованная эпилепсия (разное лечение), психогенные приступы.
Прогноз
Обычно благоприятный. У значительной части пациентов приступы возникают только при интенсивной световой стимуляции, у многих отмечаются всего 1-2 приступа в течение всей жизни, несмотря на наличие провоцирующих факторов и отсутствие лечения. Более внимательно следует отнестись к пациентам, у которых возникают также и спонтанные приступы, им может потребоваться медикаментозная терапия.
Лечение
Минимизировать провоцирующие факторы. Если требуется АЭП терапия – вальпроаты и леветирацетам, также ламотриджин, карбамазепин или клобазам.
The educational kit on epilepsies
The epileptic syndromes
By C. P. Panayiotopoulos
The Educational Kit on Epilepsies was produced through an unrestricted educational grant from UCB Pharma SA.
UCB Pharma SA assumes no responsibility of the views expressed and recommended treatments in these volumes.
Originally published by MEDICINAE
21 Cave Street, Oxford OX4 1BA
First published 2006 and reprinted in 2007
Reviewed and revised June 2008 by Steven C. Schachter, MD
Детские затылочные эпилепсии составляют гетерогенную группу (Covanis et al., 2005). Наиболее часто встречающийся тип — это идиопатическая и доброкачественная эпилепсия, впервые описанная Panayiotopoulos и в настоящее время называющаяся затылочная эпилепсия типа Панайотопулоса или синдром Панайотопулоса. Заболевание в этих случаях начинается у нормально развивающихся детей в промежутке между 1 и 10 годами жизни, с максимумом около 3-4 лет. Обычно припадки возникают нечасто и, в основном, случаются во время сна.
Они проявляются, главным образом, отклонением глаз и расстройствами понимания, которые трудно оценить во время сна. Часто выражена вегетативная симптоматика, особенно рвота. Чаще всего припадки длятся несколько минут, хотя в части случаев, до 44% (Ferrie et al., 1997), они могут длиться от нескольких минут до нескольких часов с рвотой, потерей сознания и девиацией глаз. В последнем случае они могут быть ошибочно приняты за кому токсического или метаболического генеза (Panayiotopoulos 1989, 2000; Kivity и Lerman, 1992) или могут имитировать острый живот. Длительные припадки могут завершиться продолжительными односторонними судорогами. Атипичные случаи могут манифестировать как синкопальные состояния, их бывает трудно отличить от сердечных или сосудистых расстройств.
Симптомы зрительных расстройств возникают редко. Часто бывает постиктальная головная боль. На интериктальной ЭЭГ могут выявляться различные типы спайков, обычно локализованные или преобладающие в затылочной области, одно- или двусторонние, но иногда ЭЭГ бывает нормальной. При зрительной фиксации эта активность снижается или исчезает. Такие случаи, возможно, составляют наиболее часто встречающийся доброкачественный эпилептический синдром в возрасте до пяти лет и, вероятно, связаны с роландической эпилепсией. Границы данного синдрома полностью не определены: некоторые атипичные клинические проявления, такие как синкопальные состояния и продолжительная кома, могут вводить в заблуждение; на ЭЭГ локализация и вид пароксизмальной активности может сильно варьировать с преобладанием экстраокципитальных пароксизмов в 10% случаев (Panayiotopoulos, 2002). Представляется, что эта форма часто протекает доброкачественно, хотя есть сообщения о редких атипичных случаях (Ferrie et al., 2002; Kikumoto et al., 2006).
Другой синдром, известный как доброкачественная затылочная эпилепсия типа Гасто, характеризуется наличием парциальных припадков с преимущественно зрительными симптомами, связанными с наличием над одной или обеими затылочными областями продолжительной более или менее ритмичной спайк-волновой активности, которая прекращается или значительно снижается при открывании глаз. Заболевание дебютирует обычно в возрасте 3-9 лет (Gobbi и Guerrini, 1998). Зрительные припадки могут проявляться негативными феноменами (транзиторная потеря зрения) или позитивной симптоматикой, в основном, простыми зрительными галлюцинациями геометрических форм и/или цвета (Aso et al., 1987; Thomas et al., 2003).
Также могут развиваться или быть единственным симптомом невизуальные припадки (Panayiotopoulos et al., 1989). Постиктальная головная боль, часто мигренозная, возникает примерно в трети случаев доброкачественной затылочной эпилепсии.
Идиопатические парциальные фотосенситивные эпилепсии представляют собой редкий третий тип затылочной эпилепсии (Guerrini et al., 1994b, 1995). Припадки характеризуются зрительными феноменами. Нередко они длительные, с медленно прогрессирующими зрительными нарушениями. Синдром обычно хорошо поддается лечению и имеет благоприятный исход.э

- Вернуться в оглавление раздела "Неврология."
Редактор: Искандер Милевски. Дата публикации: 4.1.2019
Ермоленко Н.А. 1 , Бучнева И.А. 1
1 Воронежская областная детская клиническая больница №1
В Классификации эпилептических приступов, эпилептических синдромов и схожих состояний, Нью-Дели, 1989г., отдельно выделялись идиопатические эпилепсии, связанные с локализацией, однако некоторые формы, которые в настоящем рассматриваются как ИФЭ, были отнесены к другим рубрикам - например, к генерализованным эпилепсиям или не были включены в Классификацию вовсе. В настоящее время произошел пересмотр таксономического положения отдельных форм эпилепсий, описаны новые формы заболевания. В связи с этим, в частности, изменились и взгляды на терапию. В проекте новой Классификации (Новой Диагностической Схеме) выделены 3 группы ИФЭ: идиопатические фокальные эпилепсии младенчества и детства, семейные (аутосомно-доминантные) фокальные эпилепсии и рефлекторные эпилепсии.
Идиопатические фокальные эпилепсии младенчества и детства
Доброкачественные младенческие приступы (несемейные) Доброкачественная эпилепсия детского возраста с центрально-темпоральными спайками
Доброкачественная затылочная эпилепсия детского возраста с ранним дебютом (Панайотопоулоса тип)
Детская затылочная эпилепсия с поздним дебютом (Гасто тип)
Семейные (аутосомно-доминантные) фокальные эпилепсии
Доброкачественные семейные неонатальные приступы Доброкачественные семейные младенческие приступы Аутосомно-доминантная ночная лобная эпилепсия Семейная височная эпилепсия
Семейная фокальная эпилепсия с вариабельными фокусами
Терапия лобной, височной и эпилепсии с вариабельным фокусом включает антиэпилептические препараты (АЭП) первой линии: карбамазепин, окскарбазепин, топирамат, леветирацетам, ламотриджин; и АЭП второй линии: лакосамид (с 16 лет), вальпроат и другие.
В предложениях по терминологии и классификации ILAE включено пять эпилептических синдромов (не считая неонатальных эпилепсий), относящихся к доброкачественным фокальным эпилепсиям: доброкачественные младенческие приступы, доброкачественные семейные младенческие приступы (синдром Ватанабе-Виджевано), доброкачественная эпилепсия детского возраста с центрально-темпоральными спайками - роландическая эпилепсия (РЭ), доброкачественная затылочная эпилепсия детского возраста с ранним дебютом (синдром Панайотопоулоса), детская затылочная эпилепсия с поздним дебютом (тип Гасто) (Engel J. Jr.,2001) .
В настоящее время широко дискутируются вопросы назначения и эффективности антиэпилептической терапии (АЭТ) при этих формах эпилепсии. Некоторые авторы считают, что противосудорожная терапия при ДФЭ обычно эффективна и должна быть назначена как можно раньше (Panayiotopoulos C.P, 1999). Исключением является синдром Панайотопоулоса. Несмотря на высокий риск развития вегетативного эпилептического статуса при синдроме Панайотопоулоса, у 27% пациентов имеют один единственный приступ, у 47% больных отмечается от 2 до 5 приступов и только у 5% - более 10 приступов. Тем не менее, активный приступный период достаточно кратковременный, у 79% пациентов через 1-2 года наступает спонтанная ремиссия без лечения, у 1/3 больных отмечается эволюция в другой вид эпилепсии, в 13% случаев наблюдается эволюция в роландическую эпилепсию (Panayiotopoulos C.P., 2005).
У части пациентов с РЭ с редкими ночными приступами, протекающими без потери сознания, АЭТ также может не назначаться (Panayiotopoulos C.P., 1999, 2005).
Выздоровление при РЭ наступает независимо от лечения, в возрасте от 9 до 12 лет, когда прекращаются приступы, затем, в среднем через 2 года нормализуется ЭЭГ. По мнению других авторов в лечении пациентов с РЭ необходимо добиваться не только клинической, но и электроэнцефалографической ремиссии, что требует назначения АЭТ длительно (не менее 36 месяцев) (Deonna T.,2000).
Несмотря на то, что при РЭ с возрастом всегда наступает выздоровление, приступы у небольшой части пациентов могут быть резистентными к АЭТ (Dalla Bernardina B. и соавт., 2002; Lerman P. И соавт., 1975; Loiseau P., 1993). Также не исключено возобновление приступов после наступления клинической ремиссии на фоне АЭТ (Fejerman N., Caraballo R.,2007). Рецидивы у взрослых крайне редки и возникают в специфических провоцирующих ситуациях (алкоголь, бессонные ночи).
Однако, по мнению ряда авторов, ответ на АЭТ не может рассматриваться как один из критериев доброкачественности ДФЭ, так как отмечено длительное персистирование эпилептических приступов у некоторых пациентов вплоть до возникновения спонтанной ремиссии, несмотря на проводимое лечение (Dalla Bernardina B. и соавт., 2002; Lerman P. и соавт. 1975; Loiseau P., 1993). Таким образом, у некоторых больных течение ДФЭ может принимать тяжелый и фармакорезистентный характер в отношении приступов (Мухин К.Ю.,2010).
F. Vigevano (2001) при лечении доброкачественных младенческих приступов рекомендует назначать вальпроаты и барбитураты. K. Watanabe (1987) применял карбамазепин. Препаратами первой очереди при лечении вегетативного эпилептического статуса у больных с синдромом Панайотопоулоса являются бензодиазепины (Roger J. и соавт.,2005).
Несмотря на отсутствие объективного преимущества какого-либо препарата при проведении монотерапии у пациентов с ДФЭ чаще всего предпочтение отдается карбамазепину (Engel J. Jr., 2001). Однако, начиная с 1999г., появляются публикации о негативном влиянии карбамазепина на когнитивные функции, о том, что он может приводить к появлению негативного миоклонуса, атипичных абсансов, атонических приступов у пациентов с РЭ (Carabolo R. и соавт., 2005; Lerman P. и соавт.,1975; Loiseau P., 1993). P.S. Dimova и соавт. (2002) при обследовании 66 детей с РЭ у 6 из них обнаружили генерализованную спайк-волновую активность и наличие абсансов, считая это осложнением терапии карбамазепином.
Рис.1 Сравнительная эффективность стартовой монотерапии АЭП у пациентов с ДФЭ (n=106) с достижением клинической ремиссии на первом году лечения 
В работе D.Corda и соавт. (2001) оценивалась частота аггравации у 98 пациентов с РЭ на фоне приема карбамазепина, фенобарбитала, вальпроата и бензодиазепинов. Аггравация была отмечена в 1 из 40 случаев на фоне терапии карбамазепином, в 1 из 12 случаев на фоне терапии фенобарбиталом; вальпроаты и бензодиазепины аггравации не вызвали. Вальпроаты в настоящее время - одни из препаратов первого ряда для лечения РЭ.
Из АЭТ нового поколения в случаях невосприимчивости к вальпроату в последнее время успешно применяется леветирацетам (Garcia C. и совт., 2009).
Л.Р. Зенков и соавт. (2007) предлагают применять для коррекции психических и поведенческие расстройств при идиопатических эпилептиформных фокальных разрядах вальпроат, ламотриджин, леветирацетам.
Об эффективности ламотриджина при РЭ сообщалось T.F. Barron и соавт. (2008). Препарат был рекомендован для первой монотерапии. Однако были единичные сообщения и об аггравации приступов и появлении негативного миоклонуса на фоне терапии ламотриджином, аналогично парадоксальной реакции на карбамазепин.
При атипичной эволюции К.Ю. Мухин и соавт. (2000) рекомендуют монотерапию высокими дозами вальпроатом (70-100 мг/кг в сутки), при неэффективности -в комбинации с ламотриджином. Однако дозы свыше 40-50 мг/кг массы тела в сутки обладают высокой токсичностью и требуют обязательного контроля за побочными эффектами, в первую очередь -уровнем тромбоцитов и печеночных ферментов.
D. Bernardina (1989) рекомендует последовательно следующие препараты: бензодиазепины (в первую очередь клобазам), сультиам, этосуксимид, внутривенный иммуноглобулин, гидрокортизон.
Этосуксимид назначается в комбинации с препаратом широкого спектра действия и был эффективен не только в отношении атипичных абсансов, но и негативного миоклонуса В работе И.А. Бучневой (2010) наиболее эффективными схемами для лечения продолженной эпилептиформной активности в медленном сне оказались комбинации вальпроата и этосуксимида или вальпроата и леветирацетама.
При проведении нами проспективного исследования 106 пациентов с ДФЭ (59 (56%) мальчиков, 47 (44%) девочек; средний возраст 7,5±0,7 лет) в течение 3-5 лет АЭТ не проводилась в течение всего периода наблюдения 19 пациентам (18%) в связи с редкостью эпилептических приступов (рис.1).
Была показана высокая эффективность стартовой монотерапии с достижением клинической ремиссия от 75%-100% пациентов в зависимости от нозологии. Исключение составили пациенты с РЭ, которые достигли клинической ремиссии на фоне стартовой монотерапии только в 54% случаев.
Наиболее эффективными АЭП в стартовой монотерапии был вальпроат в дозах 20-40 мг/кг в сутки (максимальная доза 50 мг/кг в сутки), значительно более низкую эффективность показали топирамат (3-5 мг/кг/сут) и карбамазепины (10-20 мг/кг/сут).
На фоне терапии в течение первого года мы наблюдали атипичную эволюцию (АЭ) у 13 пациентов (2 пациента с СП; 11 пациентов с РЭ) с присоединением полиморфных эпилептических приступов (негативного миоклонуса, атипичных абсансов, ГСП) и появлением продолженной диффузной эпилептиформной активности на ЭЭГ с высоким спайк-волновым индексом (СПИ) во сне (более 50%). У 3 пациентов АЭ отмечалась спонтанно, у 7 - на фоне приема карбамазепина и у 3 -на фоне приема вальпроата. Таким образом, среди всех пациентов, которым был назначен вальпроат (n =68), АЭ отмечалась в 4,4% случаев (n=3), что является результатом случайной связи, и носит характер спонтанной эволюции, в отличие от фармакоин-дуцированной эволюции на фоне приема карбамазепина (n=17), которая наблюдалась в 41% (n=7) случаев. Дуотерапия назначалась 17%-25% пациентов, которые не достигли клинической ремиссии на стартовой монотерапии или имели высокий индекс эпилепти-формных разрядов на ЭЭГ (более 30%). Назначение трех АЭП проводилось у 20% пациентов с РЭ и 7% пациентов с синдромом Панайотополуса. Через 3 года терапии клинической ремиссии достигли все пациенты (n=106).
Наиболее эффективными были комбинации вальпроата с леветирацетамом (20-40 мг/кг/сут) и/или суксилепом (15-35мг/кг/сут). Достижение клинико-ЭЭГ ремиссии отмечалось достоверно (p
Идиопатическая эпилепсия отличается от других форм тем, что отсутствуют признаки органического поражения головного мозга, и она является самостоятельным заболеванием. Чаще всего причиной является генетическая предрасположенность. Генерализованная форма отличается от локализованной более благоприятным прогнозом и лучше поддается лечению. Эпилепсия чаще встречается в развитых странах; однако такая статистика может быть связана и с более высоким уровнем медицины, а также качественной и своевременной диагностикой.
Этиология и патогенез

Идиопатическая эпилепсия считается первичным заболеванием, и ее возникновение связывают с генетической предрасположенностью. Патогенез генерализованной и парциальной форм различается.
При фокальной форме идиопатической эпилепсии в коре головного мозга формируется патологический очаг возбуждения с эпилептическими клетками, которые генерируют импульсы, приводящие к возникновению приступов.
Принципы классификации
Идиопатическая эпилепсия делится на группы согласно места расположения очага: генерализованные и локализационно-обусловленные формы.
| По локализации | Формы заболевания |
|---|---|
| Генерализованные формы | С доброкачественным течением |
| Абсанс эпилепсия детская и юношеская | |
| Эпилепсия с генерализованными судорожными приступами пробуждения | |
| Фотосенситивная эпилепсия | |
| Редкие формы идиопатической эпилепсии | |
| Локализационно-обусловленные формы | Первичная эпилепсия чтения |
| Роландическая эпилепсия | |
| Эпилепсия с локализацией в затылочной области |
Виды припадков

При диагностике генерализованных форм важным остается определить тип приступа, которые преобладает в клинической картине заболевания. Это необходимо для правильного подбора медикаментозного лечения и определения динамики.
Простой абсансный приступ формируется без двигательного компонента, но чаще к нему присоединяются миоклонический, тонический или атонический компонент, либо двигательные автоматизмы.
Присоединение миоклонического компонента характеризуется ритмичными двусторонними подергиваниями мышц лица и верхних конечностей. Такие судороги можно спровоцировать гипервентиляцией и стимуляцией светом. Ритмичное вытягивание губ в трубочку, неконтролируемые сосательные движения, миоклония век чаще наблюдается при детской абсанс эпилепсии. Для этой же формы характерны и приступы с преобладанием тонического компонента: отклонение головы и туловища кзади, тоническое отведение глаз и ассиметрическое напряжение мышц.
Генерализованные припадки
А вот генерализованные приступы всегда сопровождаются аурой, потерей сознания и, как следствие, падение пациента.

При генерализованных тонико-клонических припадках эпилептическая активность поражает оба полушария. Тонические судороги проявляются в форме резкого напряжения мышц всего туловища или отдельных групп. А клонические выглядят как мелкие подергивания симметричных или несимметричных групп мышц.
После припадка, который может длиться от 1 до 5-6 минут сознание начинает восстанавливаться. Этот процесс напрямую зависит от длительности приступа: чем он длиннее, чем пациент дольше находится в бессознательном состоянии. При этом происходит полное расслабление мышц, вплоть до непроизвольного акта дефекации или мочеиспускания.
Формы заболевания у детей
Детская абсанс эпилепсия проявляется типичными генерализованными абсансными приступами без потери сознания и судорог. Чаще болеют девочки (2:1) с началом заболевания в возрасте до 9 лет.
Юношеская абсанс эпилепсия появляется у подростков на фоне полного благополучия и незаметно для него самого и его родителей. Приступы появляются редко и не столь выражены, как при детской абсанс эпилепсии. Поэтому бывает, что до прогрессирования заболевания и появления генерализованных приступов диагностики эпилепсии не проводится. Эти судороги носят клоническо-тонический характер. Прогноз для лечения благоприятный и ремиссия при оптимальной медикаментозной терапии составляет более 80%.
Также к генерализованным формам относят эпилепсию с изолированными генерализованными приступами. Клинически она проявляется как резкая потеря сознания; приступу не предшествует аура.
Сам приступ состоит из двух фаз:
- Тоническая фаза — пациент теряет сознание,
![]()
падает. Из-за тонического спазма мышц гортани из его рта вырывается стон или крик. Все тело выгибается, руки и ноги напряжены и выпрямлены. Глаза его открыты, дыхание отсутствует, а лицо бледное. Фаза длится не более 30 секунд; - Клоническая фаза. Спазм мускулатуры проходит, возвращается дыхание (хриплое). Мышцы начинают ритмично сокращаться, подергивания приобретают все более размашистый характер. В этой фазе может произойти прикусывание языка, непроизвольное мочеиспускание или дефекация. Длится она до 10 минут, а после пациент засыпает.

Чаще всего приступ провоцируют различные нарушения сна, а сам припадок формируется сразу после пробуждения. У женщин припадки могут учащаться в предменструальный период.
Редко встречающиеся формы заболевания
Фотосенситивная эпилепсия относится к идиопатическим формам с рефлекторной зависимостью. Провоцировать возникновение приступов могут просмотр телевизора или длительное время за монитором компьютера, наблюдение за мелкими, ритмично движущимися объектами, мелькание фар, цветомузыка в клубе и многое другое.
У 
пациентов возможно формирование фотосенситивной эпилепсии со спонтанными приступами. Они могут быть спровоцированы, а могут возникать на фоне полного благополучия. При осмотре у пациентов отмечается светобоязнь, слезоточивость, чувство рези в глазах, частое моргание и головная боль. Помимо основного противосудорожного лечения хороший эффект дает профилактика (избегание провоцирующих факторов). Сюда можно отнести ношение солнцезащитных очков, избегать просмотра передач с частой сменой картинки, не посещать клубы с цветомузыкой и так далее.
Диагностика
Эпилепсия на данное время является проблемой мирового масштаба. Ее диагностика должна осуществляться на ранних этапах и быть максимально точной.
Для начала необходимо выявить причину возникновения приступов, если это возможно. В том случае, когда этиология известна, то лечение строится по принципу влияния на первопричину. Для этого необходимо тщательно опросить пациента и его родственников на предмет черепно-мозговых травм, онкологических заболеваний, 
инфекционного менингита, а также выяснить возможную генетическую предрасположенность.
Ключевым методом в диагностике идиопатической эпилепсии является электроэнцефалография. Она построена на принципе определения разности электрических потенциалов между двумя точками. Для этого над головным мозгом располагают электроды, которые фиксируют электрические импульсы.
С помощью этого метода можно определить локализацию очага возбуждения (если таковой имеется), характеристику волн (они специфичны для разных типов эпилептической активности), а также изменения в межприступный период.
Помимо этого важным является осмотр неврологом. Он проверяет выраженность рефлексов, отклонение в интеллектуальной и эмоциональной сферах.
Вспомогательными методами исследования считаются МРТ и КТ, ангиография сосудов мозга, ЭХО-энцефалограмма.
Терапия
Идиопатическая эпилепсия требует длительного и продуманного лечения. Курс противосудорожный препаратов может быть назначен только после постановки окончательного диагноза. Важны не столько форма приступов, сколько форма эпилепсии.
Назначение антиконвульсивных препаратов происходит в соответствии со схемой. Придерживаются принципов монотерапии старта с малых доз. Это значит, что лечение лучше начинать с одного препарата в малой дозировке, постепенно ее увеличивая до получения требуемого терапевтического эффекта. Если он не достигается, то препарат меняют на другой и начинают опять с небольших дозировок.
В случаях резистентности к медикаментозной терапии можно прибегнуть к хирургическому лечению, стимуляции блуждающего нерва и кетогенной диете.
Показания к хирургическому вмешательству должны быть строгими: идиопатическая парциальная эпилепсия, подходящее соматическое состояние.
Кетогенная диета направлена на снижение количества приступов. Начинать ее врачи рекомендуют с голодания: обыкновенная питьевая негазированная вода. НА четвертые сутки можно добавлять в рацион пищу. Необходимо уменьшить количество углеводов, поступающих с пищей, а количество жиров увеличить.
При достижении полной ремиссии у пациентов не должны возникать приступы на протяжении одного года. Следует стремиться к нормализации показателей электроэнцефалограммы.
Прогноз зависит от того, как заболевание реагирует на проводимую терапию и регистрируется ли прогресс течения эпилепсии.
В настоящее время эпилепсия входит в пятерку наиболее распространенных неврологических заболеваний. Она нередко дебютирует уже в детском возрасте и относится к потенциально инвалидизирующим патологиям. Причем в большинстве случаев диагностируется идиопатическая форма эпилепсии. Это классический и наиболее часто встречающийся вариант заболевания, с несколькими клиническими разновидностями и вариантами течения.
Что это за болезнь

Эпилепсией называют хроническое заболевание, обусловленное патологией на уровне головного мозга и проявляющееся преимущественно приступами (припадками) судорожного и бессудорожного характера. Свойственны ей и другие, менее яркие симптомы. Изучением всех связанных с эпилептической болезнью проблем занимается особая наука эпилептология, но в повседневной клинической практике диагностику и лечение нередко проводит невролог (невропатолог).
Согласно действующей классификации, эпилепсия подразделяется на идиопатическую, симптоматическую и криптогенную. Эти неоднородные по происхождению и симптоматике состояния отличаются друг от друга характером изменений в центральной нервной системе.
Идиопатическая эпилепсия относится к самостоятельно возникающим заболеваниям. Раньше ее называли истинной, генуинной, первичной. Она характеризуется несколькими признаками:
- В головном мозге нет органических (структурных) изменений. Поэтому такой форме болезни не свойственны очерченная очаговая неврологическая симптоматика, задержка психомоторного развития или когнитивный регресс. Современные высокоточные методы нейровизуализации оказываются не информативными.
- Приводящие к припадкам нарушения носят функциональный характер и не обусловлены действием каких-либо внешних факторов или появившихся в течение жизни заболеваний.
- Склонность к раннему проявлению симптоматики. Идиопатическая эпилепсия в 70-75% случаев дебютирует в детском возрасте, с основным пиком заболеваемости у подростков в 9-14 лет. Поэтому обычно речь идет о детских и ювенильных (юношеских) формах болезни.
- Наследственная предрасположенность. В настоящее время выделено около 500 генов в соматических хромосомах, мутация в которых может стать причиной эпилепсии. Многие формы болезни характеризуется аутосомно-доминантным или рецессивным типом наследования.
- Достаточно высокая чувствительность к современным противосудорожным препаратам. Это позволяет в большинстве случаев добиться значительного урежения приступов или даже перевести заболевание в стойкую ремиссию.
На долю идиопатических типов приходится около 70-75% случаев эпилепсии с различными клиническими проявлениями, при этом преобладают ее генерализованные формы.
Что происходит в мозге при идиопатической эпилепсии
Болезнь не связана с локальной гибелью или ишемией (выраженным кислородным голоданием) нервных клеток, опухолевым ростом, внутричерепной гипертензией, пороками развития или какими-либо другими патологическими процессами. Все изменения носят функциональный характер и формируются на клеточном и биохимическом уровнях.
Согласно современным представлениям, значительная часть случаев идиопатической эпилепсии обусловлена каналопатией – дисфункцией ионных каналов в стенках нервных клеток. Такая проблема обычно обусловлена патологией одного или нескольких генов, кодирующих структуру трансмембранных белков.
Результатом подобных каналопатий становится дефект транспорта отдельных ионов (калия, натрия) или целых молекул нейромедиаторов (ГАМК, ацетилхолина и др.), что приводит к комплексу нарушений:
- изменение скорости проведения сигналов в нервной ткани, склонность к каскадному распространению пароксизмальных импульсов;
- поддержание дисбаланса между процессами торможения и возбуждения в головном мозге, с преобладанием активирующих влияний;
- создание предпосылок для чрезмерной стойкой деполяризации на поверхности нейронов (она может быть запущена выраженным ионным дисбалансом или избыточным количеством глутамата, причем в подавляющем большинстве случаев без явного провоцирующего фактора).
При идиопатической эпилепсии соседние нейроны очень легко синхронизируются друг с другом, что объясняет склонность к быстрому тотальному распространению пароксизмально возникшего гипервозбуждения. В этот процесс генерализации вовлекаются и подкорковые регулирующие структуры, что ускоряет передачу эпилептического импульса. В результате возбуждение охватывает практически весь головной мозг, провоцируя приступ с различной симптоматикой.
При этом у каждого пациента формируется собственная достаточно стабильная схема вовлечения и реакции различных мозговых структур. Это определяет характерную клиническую картину болезни, с преобладанием определенных симптомов во время приступа.
Основные проявления
Ключевое проявление эпилепсии – разнообразные приступы, которые могут комбинироваться друг с другом или существовать в изолированном виде. Они бывают судорожными (сопровождающимися неконтролируемой пароксизмальной активностью скелетных мышц) и бессудорожными, фокальными и генерализованными.
Нередко развитию приступа предшествует аура. А при серии следующих друг за другом припадков с генерализацией говорят о развитии эпилептического статуса, что чревато развитием отека головного мозга.
Судорожные приступы
При идиопатической эпилепсии возможно появление приступов любого типа. Но преобладание одного из них или характерная трансформация разных видов друг в друга позволяет дифференцировать формы болезни.
К основным вариантам припадков относят:
Для большинства идиопатических форм болезни не свойственно развитие неврологического дефицита и выпадение высших корковых функций. Физическое и интеллектуальное развитие у детей обычно не страдает, если нет осложняющих ситуацию сопутствующих заболеваний и предшествующих перинатальных вредностей.
Изменения личности по эпилептическому типу возникают нечасто, лишь при тяжело протекающих формах болезни с частыми плохо контролируемыми генерализованными приступами и эпистатусами.
Классификация
В настоящее время общепризнанной считается классификация, основы которой были приняты еще в 1981 году Международной Лигой по борьбе с эпилепсией (Киото, Япония). При этом учитывается характер имеющихся у пациента приступов, с оценкой их разновидности и склонности к генерализации.
В соответствии с таким подходом, все варианты болезни подразделяются на 2 основные группы: генерализованные и парциальные (фокальные). Внутри каждой их них имеется несколько различных заболеваний, отличающихся по клинической картине, времени проявления симптомов и прогнозу.
Выделяют несколько основных клинических разновидностей (по действующей классификации ILAE от 1989 г.):
- Детская абсансная эпилепсия – бессудорожный вариант заболевания с доказанной наследственной природой. Характеризуется частыми многократными приступами по типу абсанса у детей 4-9 лет. На нее приходится 10-17% от всех идиопатических форм.
- Ювенильная (юношеская) абсансная эпилепсия, с дебютом в подростковом возрасте. Диагностируется примерно в 12% случаев, у ⅔ пациентов дебютирует в возрасте 9-13 лет. При этом первоначально у ребенка могут возникать и типичные генерализованные судорожные приступы, а в последующем преобладают абсансы.
- Доброкачественная младенческая миоклонус-эпилепсия. Дебютирует в возрасте от 4 мес. до 3 лет с миоклонических вздрагиваний, в последующем эти приступы приобретают генерализованный характер. Чаще заболевают мальчики.
- Ювенильная миоклонус-эпилепсия или синдром Янца. Распространенность колеблется от 4 до 12%. Патология связана с дефектом генов на коротком плече 6 соматической хромосомы. В клинике преобладают миоклонические припадки, хотя они могут дополняться эпизодически возникающими абсансами и генерализованными судорожными приступами.
- Идиопатическая эпилепсия с генерализованными судорожными приступами (изолированными).
- Эпилепсия с генерализованными судорожными приступами периода пробуждения. Заболевание с доказанной генетической детерминированностью и дебютом преимущественно в 9–11-летнем возрасте. Характеризуется появлением типичных генерализованных припадков с тонико-клоническими судорогами в просоночном состоянии.
- Эпилепсия с фотосенсибилизацией. Характеризуется рефлекторным возникновением приступов в ответ на зрительную стимуляцию, с преобладанием генерализованных судорожных тонико-клонических припадков. Пик заболеваемости приходится на возраст 12-14 лет, но первичные проявления возможны вплоть до 25-летнего возраста.
Генерализованная идиопатическая эпилепсия – самый частый вариант заболевания, именно она определяет почти ⅔ случаев эпиприпадков в детском и подростковом возрасте.
Эта полиморфная группа включает несколько заболеваний.
- Доброкачественные семейные судороги у новорожденных или доброкачественная семейная неонатальная эпилепсия
Один из прогностически благоприятных вариантов заболевания, несмотря на очень ранний дебют симптоматики. Первые проявления отмечаются обычно уже на 1-й неделе жизни ребенка, в виде фокальных коротких приступов с апноэ. В последующем могут присоединяться двигательные автоматизмы или клонические подергивания. Симптоматика обычно угасает к началу 2-го года жизни. Заболевание относится к редким и имеет аутосомно-доминантный тип наследования.
- Идиопатическая парциальная эпилепсия с лобными автоматизмами
Дает о себе знать обычно в возрасте 2-8 лет. Характерна дифференцировка симптоматики в ночное и дневное время. В состоянии бодрствования у ребенка преобладают сложные парциальные и абсансоподобные приступы, а во сне появляются гемифациальные (с вовлечением половины лица) моторные припадки, которые иногда имеют склонность к вторичной генерализации. Активный период болезни длится несколько лет, после чего обычно наступает стойкая ремиссия.
- Затылочная доброкачественная эпилепсия с поздним началом (синдром Гасто)
Пик заболеваемости в 8-9 лет. Проявляется непродолжительными сложными по структуре фокальными приступами: начало припадка со зрительных галлюцинаций, которые сменяются кратковременным исчезновением зрения (корковой слепотой) и односторонними клоническими судорогами. Выход из приступа через мигренозные боли.
- Семейная височная эпилепсия
Характеризуется простыми или сложными парциальными припадками и предшествующей психосенсорной аурой. Один из вариантов болезни – приступы в виде слуховых галлюцинаций, иногда дополняющиеся вегетативными расстройствами. Дебютирует обычно в подростковом возрасте, но нередко первые ее проявления возникают лишь к 25-30 годам. Очень хорошо поддается медикаментозной терапии и не склонна к трансформации или генерализации.
В последние годы некоторыми исследователями выделяется еще одна особая форма болезни: идиопатическая фокальная эпилепсия с псевдогенерализованными приступами. У пациентов с таким диагнозом изначально возникающие фокальные приступы быстро развертываются и усложняются, что клинически выглядит как их генерализация. Но при этом ЭЭГ однозначно показывает, что происходит лишь вторичная билатеральная синхронизация, без тотального охвата всего мозга.
К редким формам заболевания относится синдром Панайотопулоса или идиопатическая затылочная эпилепсия с ранним началом, дебютирующая в возрасте от 1 до 13 лет. Характеризуется тяжело протекающими приступами с ротацией (поворотом) глаз, выраженным вегетативным компонентом и длительно сохраняющимся бессознательным состоянием. Чаще всего они начинаются в период сна, и нередко первым проявлением припадка становится не приносящая облегчения рвота.
Риск развития эпилептического статуса при синдроме Панайотопулоса очень высокий. Но заболевание в целом оценивается как прогностически благоприятное, с доброкачественным течением и сохранностью у ребенка интеллектуально-мнестических функций. В большинстве случаев приступы возникают редко, всего 1-3 раза в течение жизни, не оказывают негативного влияния на развитие ребенка и на общее состояние его здоровья.
Диагностика
Обследование включает консультацию невролога, функциональную диагностику (ЭЭГ, сомнография, суточное мониторирование) и современные методы нейровизуализации для исключения органической причины припадков.
Чаще всего для исследования головного мозга назначают МРТ или КТ, предпочтительно с ангиопрограммой и дополнительным контрастным усилением. Это позволяет исключить объемные образования, пороки развития, выраженные нарушения ликвородинамики, сосудистые изменения (в том числе врожденные мальформации) и другую патологию, способную привести к очаговой эпилептической активности. Такое обследование необходимо для дифференциальной диагностики и подтверждения идиопатической природы заболевания.
Информативность ЭЭГ может быть различной. Это зависит от периода, когда проводилось исследование, формы и тяжести течения болезни. В приступный период ЭЭГ выявляет генерализованную синхронизированную билатеральную (двустороннюю, с охватом обоих полушарий головного мозга) эпилептическую активность по типу пик-волна. Разряды короткие, обычно провоцируются гипервентиляцией и фотостимуляцией, вероятность их выявления повышается при депривации (целенаправленном лишении) сна.
В большинстве случаев в межприступную (интериктальную) фазу ЭЭГ не выявляет полипиковой и генерализованной судорожной активности, общий (основной) ритм сохранен. Функциональные признаки структурных дефектов не характерны. Исключение составляет ювенильная миоклонус-эпилепсия, при которой аномальная ЭЭГ выявляется практически постоянно у 80-95% пациентов.
При некоторых формах заболевания могут выявляться также другие аномалии: доминирующий окципитальный (затылочный) дельта-ритм, эпизодическое бифронтальное (в лобных отделах обоих полушарий) замедление ритма и др.
Как лечить
Пациентам с любыми эпилептическими припадками показана диспансеризация с регулярным контролем состояния. Лечение подбирается индивидуально неврологом или эпилептологом. Иногда требуется также участие психиатра, если клиническая картина включает психовегетативные пароксизмы, обманы восприятия (галлюцинации), нарушения поведения и когнитивные расстройства.
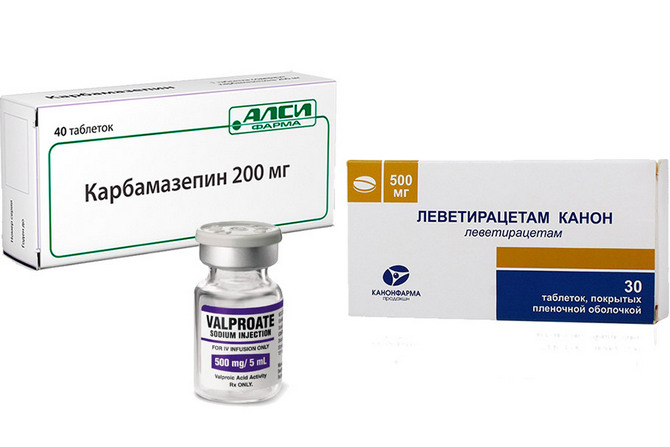
Противосудорожные препараты применяемые в терапии идиопатической эпилепсии
Основой лечения является медикаментозная терапия, с использованием антиконвульсантов (противосудорожных средств) различных групп. К наиболее употребимым препаратам относят:
- препараты вальпроевой кислоты (вальпроаты);
- Леветирацетам (Топирамат);
- Карбамазепин.
Препараты и схема их приема подбираются индивидуально, с учетом вида возникающих у пациента припадков. Лечение начинают с назначения малых доз средства 1 линии, с последующим подбором дозировки. При его недостаточной эффективности принимается решение о смене препарата или переходе на комбинированную терапию.
Противосудорожные средства принимаются постоянно, при этом необходимо регулярно контролировать картину крови и биохимические показатели работы печени. Продолжать лечение рекомендуется в течение не менее 5 лет после последнего приступа, отмена препаратов проводится постепенно, под контролем врача.
У 3-5% пациентов встречается медикаментозная резистентность – устойчивость к применяемым лекарствам, с отсутствием положительной динамики даже на комбинированной терапии достаточными дозами. В этом случае при некоторых формах заболевания может быть принято решение о целесообразности хирургического лечения, с проведением резективных или функциональных операций на головном мозге. Иногда проводят также стимуляцию блуждающего нерва.
К немедикаментозным методикам относят соблюдение режима дня, исключение провоцирующих (стимулирующих) факторов и диету. Желательно избегать потребления кофе, крепкого чая, чрезмерно острой пищи, алкоголя, тонизирующих напитков. Иногда врач дает рекомендации о переводе пациента на кетогенную диету – особую систему питания, когда ведущим источником энергии становятся жиры, а углеводы в рационе строго ограничиваются.
Прогноз
В целом идиопатические формы эпилепсии имеют относительно благоприятный прогноз. Многие из них дебютируют в детско-подростковом возрасте и в последующем постепенно угасают, иногда давая о себе знать лишь при явных провоцирующих факторах у взрослых.
Исключение составляют некоторые тяжело протекающие формы эпилепсии. Резистентность (устойчивость) к терапии нередко отмечается также у абсансных форм болезни, что требует тщательного подбора комбинации препаратов. Но даже в этом случае у ⅓ пациентов удается достичь лишь улучшения в виде значительного урежения и упрощения абсансов и исчезновения дополняющих их судорожных припадков.
Возможна также трансформация форм болезни друг в друга, несмотря на изначальное поражение разных генов. Например, эпилепсия с генерализованными приступами пробуждения может перейти в ювенильные формы абсансной или миоклонической эпилепсии. А у 30% девочек она в последующем трансформируется в катамениальную эпилепсию, при которой первично-генерализованные судорожные приступы возникают в предменструальный период.
Но в большинстве случаев удается добиться хорошего медикаментозного контроля над частотой и выраженностью приступов, избежать социально-бытовой дезадаптации пациентов и сохранить хороший темп интеллектуального развития ребенка. В стойкую ремиссию уходят 65-85% пациентов, причем большинству из них во взрослом возрасте уже не требуется постоянная поддерживающая терапия.
Читайте также:
