
Приобретенный эпилептический оперкулярный синдром
Эпилептиформный синдром - это симптомокомплекс, который выражается в эпизодических приступах судорог и бесконтрольных движений. Припадок сопровождается ухудшением самочувствия и расстройством сознания. Такие проявления нередко возникают у детей. Подобное состояние у ребенка очень пугает родителей. Однако эписиндром не имеет никакого отношения к эпилепсии. Это состояние хорошо поддается коррекции и терапии.
Что это такое
Эпилептиформный синдром (эписиндром) - это общее название судорожных припадков, которые могут быть спровоцированы нарушениями работы головного мозга. Такое отклонение не является отдельным заболеванием, это лишь одно из проявлений разных патологий.
При эписиндроме приступы судорог возникают внезапно и так же неожиданно прекращаются. Они появляются как реакция центральной нервной системы на раздражители. При этом в головном мозге формируется очаг перевозбуждения.

Припадки исчезают навсегда после излечения основной патологии. Если это нарушение возникло в детском возрасте, то оно никак не сказывается на умственном и физическом развитии ребенка.
Отличие от эпилепсии
Очень важно дифференцировать эпилептиформный синдром от эпилепсии. Это две разных патологии, имеющие похожую симптоматику. Врачи выделяют следующие основные отличия между этими двумя заболеваниями:
- Эписиндром является одним из проявлений других болезней центральной нервной системы. Эпилепсия - это отдельная патология, которая протекает в хронической форме.
- Спровоцировать появление эписиндрома могут разные заболевания. Причиной эпилепсии в большинстве случаев является наследственная предрасположенность к данной патологии.
- При эписиндроме приступы возникают эпизодически. Эпилептические припадки могут беспокоить больного на протяжение всей жизни. При отсутствии систематической терапии судороги появляются очень часто.
- Для эписиндрома нехарактерно прикусывание языка и непроизвольное мочеиспускание во время приступа. Эти признаки присущи эпилепсии.
- Перед истинным эпилептическим припадком у пациента возникает состояние ауры. Это симптомы, которые предшествуют возникновению судорог. Перед началом приступа у больного появляется дискомфорт в теле, онеменение конечностей, головокружение, зрительные нарушения, изменение восприятия запахов. При эписиндроме припадок начинается всегда неожиданно, без предшественников.
Первые признаки эпилепсии в 70% случаев появляются еще в детстве. При длительном течении патологии у больного возникают нарушения психики. Для эпилептиков характерны частые перепады настроения, депрессии, ухудшение памяти и когнитивной функции. Эписиндром может развиваться как у детей, так и у взрослых. Он не сопровождается психическими отклонениями.
Этиология
Причины эпилептиформного синдрома у взрослых и детей несколько отличаются. Данная патология у ребенка чаще всего имеет врожденный характер. Ее вызывают различные неблагоприятные факторы, воздействующие на плод во внутриутробный период:
- инфекционные болезни у матери во время беременности;
- гипоксия плода;
- травмы при родах.
В редких случаях у детей отмечается приобретенный эписиндром. Судорожный приступ может возникнуть на фоне высокой температуры (более +40 градусов) или при недостатке в организме микроэлементов (калия, натрия).
У взрослых людей эписиндром чаще всего бывает приобретенным. Его могут спровоцировать следующие патологии:
- инфекции головного мозга (энцефалит, менингит);
- травмы черепа;
- демиелинизирующие патологии (рассеянный склероз и др.);
- опухоли мозга;
- геморрагический инсульт;
- нарушение функции паращитовидных желез;
- обильная потеря крови;
- отравление тяжелыми металлами и седативными лекарственными средствами;
- гипоксия вследствие утопления или удушения.
Нередко судорожные приступы появляются у людей, злоупотребляющих спиртным. Эписиндром развивается не только у хронических алкоголиков. Иногда для возникновения припадка достаточно однократного употребления чрезмерного количества спиртосодержащих напитков.
Код по МКБ
Международная классификация болезней рассматривает эписиндром как симптоматическую эпилепсию. Эта патология входит в группу заболеваний, сопровождающихся приступами судорог. Они значатся под шифром G40. Полный код эпилептиформного синдрома по МКБ-10 - G40.2.
Симптоматика
Эта патология может протекать с разнообразной симптоматикой. Проявления эпилептиформного синдрома зависят от локализации поражения головного мозга. Если очаг возбуждения возникает в лобных долях, то во время приступа появляются следующие симптомы:
- вытягивание рук и ног;
- резкое напряжение мышц всего тела;
- болезненный спазм жевательной и мимической мускулатуры;
- закатывание глаз;
- вытекание слюны изо рта.
Если зона поражения расположена в височной части мозга, то характерны следующие проявления:
- спутанность сознания;
- раздражительность или приподнятое настроение;
- боли в животе;
- лихорадка;
- тошнота и рвота;
- слуховые и зрительные галлюцинации.
Для поражения теменной части характерна преимущественно неврологическая симптоматика:
- онемение конечностей;
- расстройство координации движений;
- сильное головокружение;
- фиксация взгляда на одной точке;
- потеря пространственной ориентации;
- обморок.
При любой локализации очага возбуждения приступ сопровождается нарушением сознания. После окончания припадка пациент ничего не помнит и не может рассказать о своем состоянии.

Довольно часто такие припадки носят единичный характер. Если же приступы возникают систематически, то врачи диагностируют эпилептический статус.
Особенности эписиндрома в детском возрасте
Эпилептиформный синдром у детей в возрасте до 1 года протекает с ярко выраженной симптоматикой. Это связано с тем, что у младенцев центральная нервная система еще не до конца сформирована. Приступ у грудничков сопровождается следующими проявлениями:
- В начале припадка появляется сильное сокращение мышц всего тела. Происходит остановка дыхания.
- Ребенок плотно прижимает руки к грудной клетке.
- У младенца выбухает родничок.
- Мускулатура резко напряжена, а нижние конечности вытянуты.
- Малыш запрокидывает голову или совершает ритмичные кивки.
- Довольно часто приступ сопровождается рвотой и выделением пены изо рта.
Эпилептиформный синдром в более старшем возрасте сопровождается судорогами лица, которые затем переходят на все тело. Дети старше 2 лет могут внезапно просыпаться и ходить по комнате в бессознательном состоянии. При этом у них отсутствует реакция на какие-либо раздражители.

Диагностика
Необходимо отличить эписиндром от истинной эпилепсии. Поэтому очень важно провести точную дифференциальную диагностику.
Пациентам назначают МРТ головного мозга. Это обследование помогает выявить этиологию эпилептиформного синдрома. Очаги глиоза на снимке указывают на повреждение нейронов вследствие травмы или инсульта. Глиозными изменениями врачи называют разрастание вспомогательных клеток головного мозга. Обычно это отмечается после гибели нейронов.
Важным методом дифференциальной диагностики является электроэнцефалограмма. При эписиндроме ЭЭГ может не показывать патологических изменений. Ведь очаги возбуждения в головном мозге появляются только перед приступом. При эпилепсии электрическая активность коры мозга повышена постоянно.
Методы терапии
Эписиндром исчезает только после устранения его причины. Поэтому необходимо пройти курс терапии основного заболевания. Одновременно проводят симптоматическое лечение эпилептиформного синдрома. Назначают следующие группы препаратов:
- Противосудорожные лекарства: "Карбамазепин", "Ламотриджин", "Депакин", "Конвулекс". Эти средства купируют судороги и уменьшают частоту припадков.
- Седативные препараты: "Фенибут", "Феназепам", "Элениум", "Атаракс". Эти средства успокаивают очаг возбуждения в головном мозге и расслабляют мышцы.
В качестве дополнительного метода лечения используют фитотерапию. Пациентам рекомендуется принимать отвары из фиалки, липы, пижмы, багульника. Эти лекарственные растения успокаивают центральную нервную систему.
При эпилептиформном синдроме пациентам показана диета. Из рациона следует исключить острые и соленые блюда, а также ограничить количество углеводов и белков. Такие продукты могут спровоцировать приступ. Рекомендуется уменьшить количество потребляемой жидкости.

В большинстве случаев эписиндром поддается консервативной терапии. Оперативное лечение применяется редко. Нейрохирургические операции проводят только при наличии новообразований в головном мозге.
Прогноз
Данное нарушение является лишь симптомом других заболеваний. Поэтому прогноз при эпилептиформном синдроме будет полностью зависеть от характера основной патологии. Если данное состояние спровоцировано инфекциями, то такие заболевания хорошо поддаются терапии антибиотиками. Если же причиной эписиндрома стала черепно-мозговая травма, рассеянный склероз или инсульт, то лечение может быть довольно продолжительным.
В целом эпилептиформный синдром имеет благоприятный прогноз. Если это нарушение возникло в детском возрасте, то к пубертатному периоду приступы обычно исчезают. Эписиндром не приводит к интеллектуальным нарушениям и не сказывается на умственном развитии ребенка. В большинстве случаев припадки бесследно проходят к 14-15 годам.

Детский невролог
26 лет медицинского стажа
Кандидат медицинских наук, доцент
Омельяненко А.А., Евтушенко С.К.
Идиопатические эпилепсии у детей представляют более половины всех случаев эпилепсии и предполагают отсутствие выявляемого морфологического субстрата, отсутствие неврологического дефицита в межприступный период. Они наиболее вероятно являются генетически обусловленными синдромами, демонстрируют возрастзависимость и доброкачественное течение в большинстве случаев. Список этих эпилептических синдромов постепенно расширяется [3, 10].
"Классическим" представителем этой группы эпилепсий является роландическая эпилепсия (РЭ) или доброкачественная детская эпилепсия с центротемпоральными спайками. Она представляет одну из наиболее частых форм эпилепсии детского возраста и составляет до четверти случаев эпилепсии в возрасте от 5 до 14 лет. Распространенность ее колеблется от 7.1 до 21 на 100000 детей до 15 лет. Истинная частота этого заболевания может быть выше, так как не все случаи диагностируются.
Простые фокальные приступы составляют основную часть приступов при РЭ и наблюдаются у 70-80% больных. Наиболее типичным является начало приступа с соматосенсорной ауры: ощущения покалывания, онемения, "прохождения электрического тока" в области глотки, языка, десен с одной стороны. На этом пароксизм может закончиться или после ауры развивается фокальный моторный приступ.
Возможные следующие варианты приступов:
гемифациальные – односторонние тонические, клонические или тонико-клонические судороги мышц лица;
фарингооральные – односторонние судороги губы, языка, глотки, гортани, которые часто соединяются с анартрией и гиперсаливацией.
У 20% больные судороги могут распространяться с лица на гомолатеральную руку (брахиофациальные приступы), и приблизительно в 8% случаев в процесс вовлекается и нога (гемиконвульсивные приступы). Приступы могут изменять свою сторонность. Вторично генерализованные судорожные приступы отмечаются у 20-25% больных РЭ. Они наиболее характерны для младших детей и нередко являются дебютным симптомом.
Продолжительность приступов, как правило, небольшая – от нескольких секунд до 2-3 мин. У 11-22% больных продолжительность пароксизмов превышает 10-15 мин. В единичных случаях отмечаются тяжелые продолжительные приступы, которые заканчиваются преходящим послеприступным гемипарезом (паралич Тодда). Частота приступов также обычно невысокая – в среднем 2-4 раза в год.
Характерным межприступным электрографическим коррелятом являются отчетливые высокоамплитудные пики с последующей выраженной медленной волной появляющиеся по одиночке или сериями в центрально-средневисочной (роландической) области (Т3 и С3 или Т4 и С4) с формированием диполя в лобных отведениях (Рис. 1-2). Количество роландических пиков нарастает во время медленного сна. Специфичность этого ЭЭГ-паттерна, конечно, не абсолютная, но очень высокая и может сравниться со специфичностью генерализованных комплексов пик-волна 3 Гц для типичных абсансов.
Диагностика роландической эпилепсии основана на сочетании клинических проявлений, нормального психоневрологического статуса, нормальной картины мозга по данным нейровизуализации и характерного ЭЭГ-паттерна.
Критерии диагностики роландической эпилепсии (Loiseau, Duche 1989):
- Дебют в возрасте от 3 до 13 лет;
- Нормальные психический и неврологический статус до начала заболевания;
- Парциальные моторные приступы, часто с соматосенсорной аурой, провоцируемые сном;
- Очаг пиков в центротемпоральной (роландической) области с нормальной фоновой активностью;
- Спонтанное выздоровление в юношеском возрасте.
За последние 10 лет под наблюдением в нашей клинике находилось 743 ребенка с электрографической картиной роландической эпилепсии. Различные варианты вышеуказанных клинических "девиаций" были выявлены у 150 детей (20,2%). У 2 детей с типичным "роландическим" ЭЭГ-паттерном были совершенно нетипичные приступы – 1 ребенок испытывал ночные гемидизестезии до 5 минут длительностью, а другой – редкие короткие пароксизмы болей в животе во время сна. В обоих случаях заболевание завершилось ремиссией и исчезновением интериктальных разрядов на ЭЭГ.
К атипичным вариантам РЭ можно отнести случаи, когда этот синдром развивается на фоне заведомо поврежденного мозга. В нашей практике имеют место 13 случаев типичной электроклинической картины РЭ у детей с детским церебральным параличом.
Изменения ЭЭГ у наших больных с клинической картиной РЭ были типичными лишь в 63% случаев. У остальных имели место те или иные вариации ЭЭГ-паттерна – максимум амплитуды и электронегативности не в среднем височном отведении; сочетание с пиками иной локализации (чаще всего затылочными и центросаггитальными); пики исключительно в центросаггитальном отведении; сочетание с генерализованными разрядами комплексов пик-волна или полипиков; фо-тосенситивность; записи с очень обильной эпилептической активностью, которая могла бы соот-ветствовать критериям электрического эпилептического статуса (рис. 3-6) 10.
Характеристики ЭЭГ у каждого отдельного больного могут значительно изменяться со временем – пики могут исчезать совсем, вновь появляться, менять локализацию, учащаться до непрерывных и снова исчезать, чтобы возобновиться при следующем обследовании, а затем исчезнуть навсегда. Такую нестабильность электрографических проявлений мы рассматриваем как один признаков идиопатического характера эпилепсии. Реже возможна относительная стабильность интериктальной активности на протяжении всего периода заболевания.
Традиционное представление о доброкачественности и превосходном прогнозе идиопатических фокальных эпилепсий детства, представителем которых является и роландическая эпилепсия, в последнее время значительно пошатнулось. Концепция известного эпилептолога Panayiotopoulos "доброкачественной детской предрасположенности к приступам" [4] не может быть применима ко всем случаям РЭ. При РЭ возможны не только частые, тяжелые и фармакорезистентные приступы, но и сочетание с постоянными или преходящими поведенческими, когнитивными расстройствами, нарушениями речи и другими вариантами психоневрологического дефицита, которые для "классической" РЭ не характерны.
Такие формы в современной литературе объединяются в группу "роландическая эпилепсия плюс" [1-3, 5-8]:
- Роландическая эпилепсия с преходящими когнитивными нарушениями в активной фазе
- Статус роландической эпилепсии
- Приобретенный эпилептический оперкулярный синдром (оперкулярный статус)
- Роландическая эпилепсия с негативным миоклонусом
К первой категории указанных вариантов относится роландическая эпилепсия, протекающая с преходящими когнитивными расстройствами, которые возникают в период наибольшей активности болезни и прямо коррелируют с выраженностью эпилептической активности на ЭЭГ. Обычно клинически выявляемые нарушения возникают при частоте пиков более 10 в минуту, а нормализация происходит при частоте разрядов менее 5 в минуту. Для выявления таких расстройств может понадобиться нейропсихологическое тестирование или исследование когнитивных вызванных потенциалов.
Статус роландической эпилепсии представлен частыми роландическими моторными приступами с диз- или анартрией, слюнотечением, оромоторной диспраксией, гемифациальными судорогами и атоническими кивками головы в рамках негативного миоклонуса. Эпизоды негативного миоклонуса могут вовлекать руки и возникать так часто, буквально при каждом разряде, что возникают трудности в манипуляции руками и это может приводить к тому, что ребенок намеренно избегает пользоваться вовлекаемой в разряды рукой. Интериктальная ЭЭГ представлена у таких детей частыми билатеральными, нередко синхронными, разрядами комплексов пик-волна или острая медленная волна с максимумом в роландической области и с преобладанием над одним из полушарий (рис. 7-8). Мы наблюдали 2 пациентов с РЭ с периодами выраженного негативного миоклонуса, в который вовлекались контралатеральные разрядам половина лица и рука. В одном из этих случаев также имела место врожденная гипоплазия обеих почек, пациент находился на диализе и антиконвульсантная терапия не проводилась. Во втором случае отмечалась фармакорезистентность и только кортикостероиды и внутривенные инфузии иммуноглобулина приводили к краткосрочным ремиссиям.
Приобретенный эпилептический оперкулярный синдром или оперкулярный статус клинически характеризуется длительными, но отчетливо флуктуирующими эпизодами псевдобульбарного паралича с гиперсаливацией и слюнотечением, оромоторной диспраксией. Длительность каждого такого эпизода может составлять от нескольких часов до нескольких месяцев. Типичные роландические приступы в этот период не возникают, но могут быть выявлены слабо выраженные пози-тивные или негативные периоральные или фациальные миоклонии. Запись ЭЭГ в бодрствовании выявляет обильную двустороннюю роландическую эпилептическую активность не достигающую, однако, степени электрического эпилептического статуса. В периоды ухудшений может развиваться и электрический эпилептический статус сна. Мы наблюдали 3 детей с типичными орофациальными приступами и обильными "роландическими" разрядами в дебюте с последующим присоединением длительных периодов выраженного слюнотечения, умеренных дизартрии, нарушений глотания и когнитивных расстройств. Во всех случаях антиконвульсантная терапия приводила к купированию этого состояния.
В дебюте заболевания клиническая картина и ЭЭГ-паттерны могут выглядеть совершенно безобидно. Однако ее течение может изменяться и через несколько месяцев или лет состояние ребенка ухудшается - приступы учащаются, присоединяются другие их виды, резко усиливается эпилептическая активность и обычная РЭ может трансформироваться в один из вариантов из значительной группы "расстройств ассоциированных с роландической эпилепсией" [2, 6-9].
Эта группа представлена различными синдромами эпилептической энцефалопатии с роландическим ЭЭГ-паттерном, электрическим эпилептическим статусом сна и/или бодрствования, вариабельными комбинациями приступов, включая роландические - атипичная доброкачественная фокальная эпилепсия (с-м псевдоЛеннокса), с-м Патри, приобретенный эпилептический лобный синдром, с-м Ландау-Клеффнера и варианты аутистических эпилептиформных расстройств (дезинтегративное расстройство, регрессивное аутистическое расстройство) [1, 2, 6-9]. Среди наших пациентов с "роландическим" ЭЭГ-паттерном такие варианты были зарегистрированы в 57 случаях (7,8%). Течение заболевания у этих детей отличалось фармакорезистентностью приступов и преобладанием когнитивных и поведенческих расстройств над собственно иктальными проявлениями.
Таким образом, можно утверждать, "классическая" роландическая эпилепсия не является монолитной нозологической формой, а представляет лишь небольшую частью спектра "роландических" эпилепсий, которые в свою очередь являются составной частью широкого круга идиопатических фокальных эпилепсий внутри которого возможны сочетанные варианты или трансформация одного синдрома в другой.
В этом континууме, в маргинальной позиции, с одной стороны находятся пациенты с роландическим ЭЭГ-паттерном без каких-либо клинических появлений, а противоположная крайность представлена тяжелыми инвалидизирующими эпилептическими энцефалопатиями. Эпилептические энцефалопатии "исходящие" из фокальных эпилепсий в своих клинических проявлениях приближаются к "генерализованным" эпилептическим энцефалопатиям, представленными синдромами Леннокса-Гасто, Драве, Дуза (рис. 9). Общими чертами для большинства эпилептических энцефалопатий являются когнитивные расстройства, атипичные абсансы, феномен негативного миоклонуса и электрический эпилептический статус.
В связи с этим, говорить о доброкачественности и хорошем прогнозе роландической эпилепсии можно только в отношении пациентов, которые точно укладываются в критерии "классической" роландической эпилепсии. К сожалению, полное соответствие этим критериями нередко может быть установлено только лишь ретроспективно, когда заболевание уже завершилось или стала очевидной тенденция его течения.
Учитывая возможность неблагоприятного или, по крайней мере, атипичного течения роландической эпилепсии на наш взгляд целесообразно отказаться от широко распространенного убеждения в ее доброкачественности, не использовать термин "доброкачественная" в формулировке диагноза и быть осторожными в прогнозе заболевания.
Список литературы:
- Datta A., Sinclair D. Benign epilepsy of childhood with rolandic spikes: Typical and atypical va-riants // Pediatr. Neurol. – 2007. – v. 36. – p. 141-145
- Gobbi G., Boni A., Filippini M. The spectrum of idiopathic rolandic epilepsy syndromes and idi-opathic occipital epilepsies: from the benign to the disabling // Epilepsia. – 2006. – v. 47(Suppl. 2). – p. 62–66
- Lundberg S., Eeg-Olofsson O. Rolandic epilepsy: a challenge in terminology and classification // European Journal of Paediatric Neurology. – 2003. – v. 7. – p. 239–241
- Panayiotopoulos C. Benign childhood partial epilepsies: benign childhood seizure susceptibility syndromes [editorial] // J Neurol Neurosurg Psychiatr. – 1993. – v. 56. – p. 2-5
- Pressler R., Robinson R., Wilson G., Binnie C. Treatment of interictal epileptiform discharges can improve behavior in children with behavioral problems and epilepsy // J Pediatr. – 2005. – v.146. – p.112-117
- Saltik S., Uluduz D., Cokar O. et al. A clinical and EEG study on idiopathic partial epilepsies with evolution into ESES spectrum disorders // Epilepsia. – 2005. - v. 46 (4). – p. 524-533
- Stephani U., Carlsson G. The Spectrum from BCECTS to LKS: The Rolandic EEG Trait—Impact on Cognition // Epilepsia. – 2006. - v. 47 (Suppl. 2). – p. 67–70
- Евтушенко С.К. О роли электрической эпилептической энцефалопатии у детей // Проблемы детской неврологии (Международный сборник научных трудов). - Минск. - 2006. - с. 76-91
- Евтушенко С.К. Электрический эпилептический статус сна и эпилептические энцефалопа-тии у детей (клиника, диагностика, лечение) // Международный неврологический журнал. – 2006. - № 1(5). – с. 62-70
- Евтушенко С.К., Омельяненко А.А. Клиническая электроэнцефалография у детей // Донецк. – 2005. – 880 с.
ИЛЛЮСТРАЦИИ:
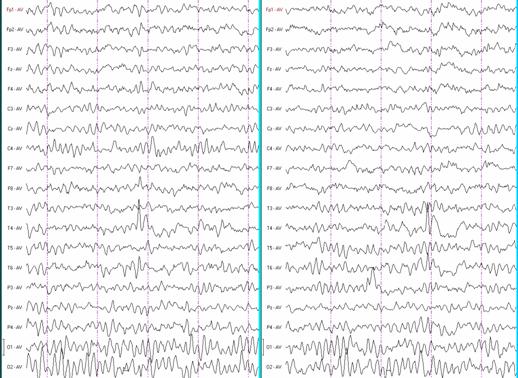
Рис. 1. Типичный ЭЭГ-паттерн роландической эпилепсии с одиночными пиками. Бодрствование. 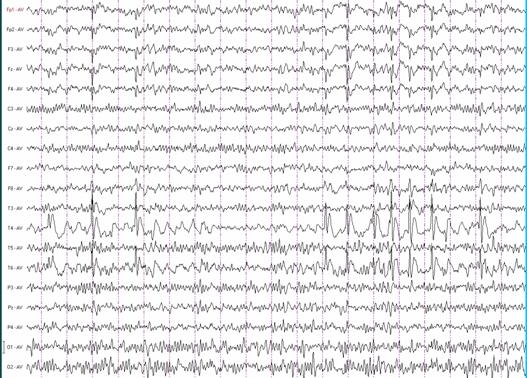
Рис. 2. Типичный роландический ЭЭГ-паттерн. Групповые пики с локальной медленной активностью. Бодрствование. 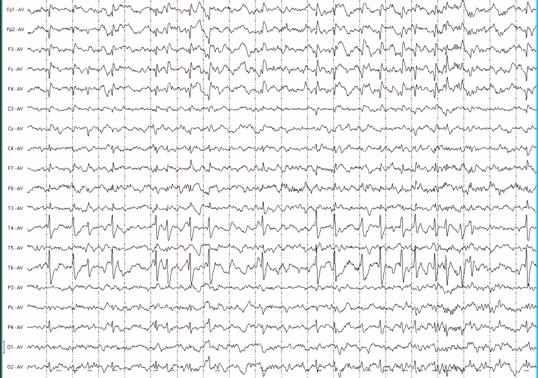
Рис. 3. Атипичный роландический ЭЭГ-паттерн с максимумом в задневисочных отделах. Сон. 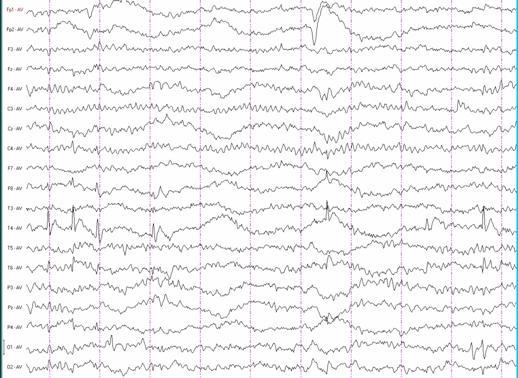
Рис. 4. Атипичный роландический ЭЭГ-паттерн. Сочетание роландических пиков с затылочными пиками. Бодрствование. 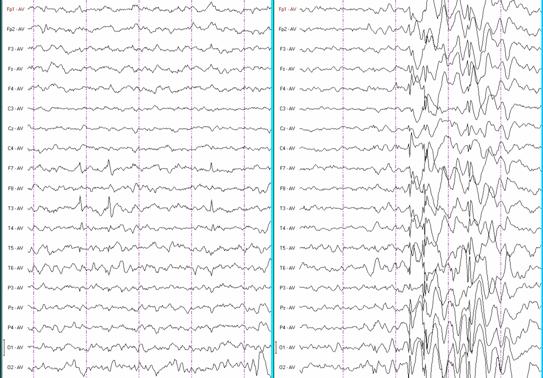
Рис. 5. Атипичный роландический ЭЭГ-паттерн. Роландические пики с генерализацией. Бодрствование. 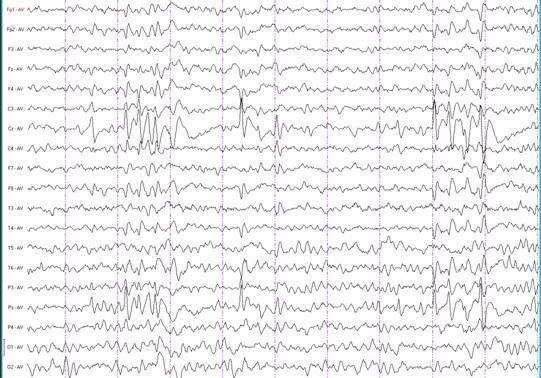
Рис. 6. Атипичный роландический ЭЭГ-паттерн. Саггитальные пики. Дремота. 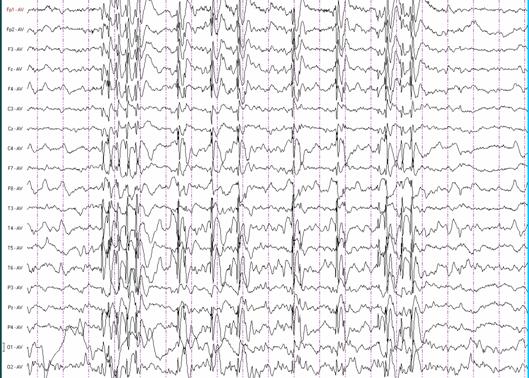
Рис. 7. Б-ной Г. 8 лет. Статус роландической эпилепсии с негативными миоклонусами. Бодрствование. 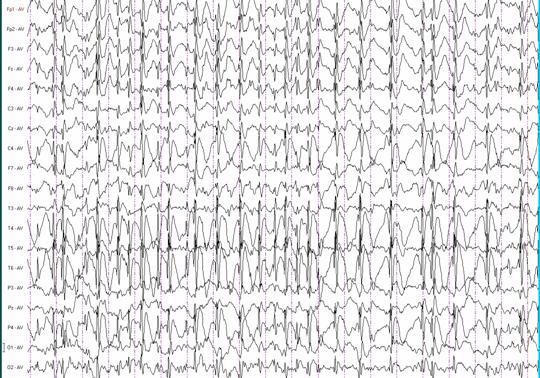
Рис. 8. Б-ной Г. 8 лет. Статус роландической эпилепсии с негативными миоклонусами. Сон. 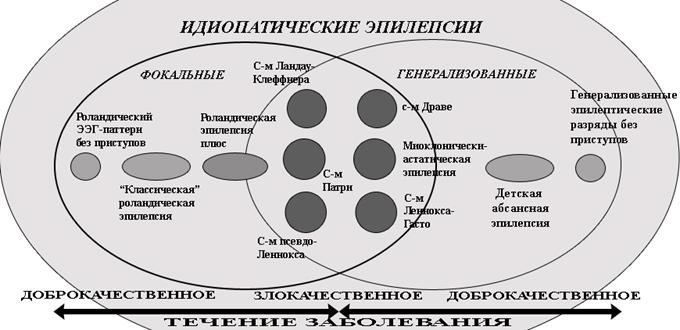
Рис. 9. Положение эпилепсий с роландическим ЭЭГ-паттерном в структуре идиопатических эпилепсий.
Вопросы и ответы
- Эпилепсия у ребенка
- Ребенок засыпает только держась за волосы мамы
- Мигрень у ребенка
- Лечение ювенильной миоклонической эпилепсии
Отзывы о специалисте
Лучший невролог, у которого мы были. Сразу смог найти подход к ребенку, не просил рассказать стишок или еще как-то искусственно вытянуть из нее слово. А сам заинтересовал рассказом о различных животных. Спокойный, внимательный и неспешный врач, с прекрасным чувством юмора. Ребенок расслабился и спокойно уснул во время энцефалограммы дневного сна.
Анатолий Анатольевич, мы с Светланкой Вам очень благодарны - и за профессионализм, и за честность, и за большое сердце. Пусть Господь благословит Вас и Ваших близких!

Cиндромы c CSWS являются функциональными расстройствами, возникающими во время активного синаптогенеза с аксональным спраутингом. Нейрональная активность или установление синаптических контактов существенно влияет на то, какие из этих синапсов останутся, а какие редуцируются до достижения 10-летнего возраста. Постоянная эпилептиформная активность во время медленного сна в период организации мозга негативно влияет на установление соответствующих нейрональных связей, нормальное его развитие и функционирование.
Сноска 1. Постоянная эпилептиформная активность во время медленного сна в период организации мозга негативно влияет на установление соответствующих нейрональных связей, нормальное его развитие и функционирование.
Эпилептические энцефалопатии, связанные с CSWS
Эпилепсия с электрическим эпилептическим статусом во время медленного сна
Согласно недавнему предложению ILAE [7] данный синдром ESES определяется как частично обратимая, возраст-зависимая детская эпилептическая энцефалопатия, характеризующаяся следующими признаками:
1) фокальные и генерализованные эпилептические приступы (унилатеральные или билатеральные клонические, тонико-клонические, абсансы, парциальные моторные, сложные парциальные приступы и эпилептические падения);
2) нейропсихологические нарушения в форме общей или избирательной регрессии когнитивных функций (исключением является приобретенная афазия, которая традиционно рассматривается отдельно);
3) двигательная недостаточность в форме атаксии, нарушении целенаправленных движений, дистонии или односторонних поражений;
4) ЭЭГ паттерны диффузных спайк-волн (унилатеральных или фокальных) занимающие не менее 85% медленного сна и персистирующие в трех записях ЭЭГ в течение, по крайней мере 1 месяца.
Необходимым условием для диагностики этого синдрома является наличие постоянных спайк-волн во время медленного сна.
Демографические данные. Синдром ESES наблюдается только у детей. Частота его составляет 0,5% от общего числа детей с эпилепсией, с преобладанием у мальчиков (63%).
Сноска 2. Синдром ESES встречается только в детском возрасте с частотой 0,5% от общего числа детей с эпилепсией с преобладанием у мальчиков (63%).
Этиология. Этиология неизвестна. Более чем у одной трети пациентов причиной ECSWS могут быть врожденные мальформации и перинатальное повреждение головного мозга [8]. При проведении нейровизуализации патологию головного мозга можно обнаружить в 60% случаев.
Сноска 3. При проведении нейровизуализации у пациентов с ECSWS патология головного мозга выявляется в 60% случаев.
Стадия относительной нормализации ЭЭГ
Через несколько лет после достижения ремиссии эпилепсии отмечается постепенное улучшение ЭЭГ. Разряды во время медленного сна становятся более короткими, менее частыми и более фрагментарными. Различимыми становятся физиологические паттерны сна. Однако на ЭЭГ сна могут длительно сохраняться редкие фокальные комплексы острых и медленных волн, даже после клинического улучшения. Нормализация ЭЭГ, если она достигается, может занимать более 15 лет [6].
Диагностика. Проведение МРТ головного мозга обязательно. Более чем у трети пациентов выявляются изменения на МРТ. Наиболее типичными являются кортикальная атрофия, порэнцефалия, нарушения кортикального развития.
Функциональная визуализация головного мозга (PET или SPECT) обычно выявляет патологию даже у пациентов с нормальной МРТ.
Электроэнцефалографическое исследование включает в себя проведение стандартной ЭЭГ, продолженного видео-ЭЭГ мониторинга или амбулаторного мониторинга. Подозрение на заболевание может возникнуть при записи ЭЭГ во время короткого сна, но для более точного определения необходима регистрация ЭЭГ на протяжении всего сна.
Дифференциальная диагностика. Дифференциальная диагностика ЕCSWS проводится с синдромом Ландау-Клеффнера [13]. При синдроме Ландау-Клеффнера доминирующим когнитивным нарушением является приобретенная афазия, эпилептические приступы могут отсутствовать, и на интериктальной ЭЭГ фокусы эпилептиформной активности локализуются преимущественно в височных отведениях, в то время как при ECSWS – преимущественно в лобных.
Следует проводить дифференциальный диагноз с синдромом Леннокса-Гасто, отличительной особенностью которого являются тонические приступы с быстрой пароксизмальной активностью на ЭЭГ, которые при ECSWS отсутствуют. Моторные приступы и ремиссии при синдроме Леннокса-Гасто крайне редки.
Прогноз. Спонтанное разрешение эпилептиформных разрядов на ЭЭГ и эпилептических приступов происходит в подростковом возрасте, что совпадает со стабилизацией или улучшением нейропсихологических и поведенческих функций [6]. Но выздоровление всегда происходит медленно и, в большинстве случаев, лишь частично. Персистирование и тяжесть резидуальных поведенческих, когнитивных и речевых нарушений зависит от возраста начала и продолжительности активной фазы эпилептиформной активности.
Приобретенная эпилептическая афазия (синдром Ландау-Клеффнера) – детское заболевание, характеризующееся ассоциацией приобретенной афазии и мультифокальных спайков и спайк-волновых разрядов на ЭЭГ.
Демографические данные. Возраст дебюта заболевания составляет 2-8 лет, с пиком в 5-7 лет. Мальчики болеют в 2 раза чаще. В специализированные центры поступают один или два новых пациента в год.
Этиология неизвестна. Наследственность отягощена по эпилептическим приступам в 12% случаев и в 5% в тех случаях, если пациенты не имеют приступов. Однако есть сообщения о найденных на биопсии аномалиях головного мозга у 3% пациентов с синдромом Ландау-Клеффнера [8].
Клиническая манифестация. Первым симптомом обычно является слуховая вербальная агнозия. Дети перестают идентифицировать звуки окружающей среды и соотносить их с предметами, что делает их похожими на больных с аутизмом. Родители замечают, что дети не реагируют даже на громкое обращение. Слуховая вербальная агнозия может прогрессировать и осложняться импрессивной и экспрессивной афазией с полной утратой произносимой речи. Диагностика, как правило, ошибочна, т.к. первоначально ставится диагноз приобретенной глухоты или элективного мутизма. Многим детям записывают аудиограмму, которая оказывается нормальной. Лингвистический дефицит может быть выявлен не сразу из-за выраженного нарушения поведения и когнитивных проблем.
Таким образом, главным когнитивным нарушением, описываемым как приобретенная афазия, является аудиторная вербальная агнозия, возникающая у изначально здоровых детей, навыки психоречевого развития и словарный запас которых соответствуют возрасту.
Начало заболевания подострое. Отмечается постепенное ухудшение речевых функций в виде изменения экспрессивной речи, появления парафазий, стереотипий, персевераций и фонологических ошибок. Ребенок начинает выражаться телеграфным стилем или очень простыми предложениями, в конечном итоге становится полностью немым и перестает реагировать на невербальные сигналы.
Один из необъяснимых признаков синдрома Ландау-Клеффнера – это флюктуация симптоматики речевых нарушений, характеризующаяся ремиссиями и ухудшениями.
Познавательные и поведенческие расстройства наблюдаются у 75% пациентов. Наиболее характерными являются дефицит внимания с гиперактивностью. В редких случаях это может прогрессировать до тяжелой расторможенности и психоза. Длительное исследование этих пациентов в динамике показало, что интеллектуальная недостаточность развивается не во всех случаях.
Эпилептические приступы наблюдаются также у 75% пациентов, но они редкие и имеют хороший прогноз. Дебют приступов наблюдается в возрасте 4-6 лет. Только у 20% пациентов приступы продолжаются после 10 лет, но до 15 лет полностью исчезают.
Приступы полиморфные: фокальные моторные, атипичные абсансы, атонические приступы с падениями, малые автоматизмы и вторично-генерализованные. Кратковременные приступы с минимальными моторными и субъективными симптомами могут быть частыми, но они трудноуловимы и могут не определяться. В 1/3 случаев отмечаются только генерализованные тонико-клонические приступы или изолированный эпилептический статус, который может возникать в возрасте 5-10 лет. Сложные парциальные приступы, характерные для лобнодолевого региона, исключены, так же как и тонические приступы.
Приступы чаще ночные, редкие, хорошо поддаются лечению и полностью исчезают в возрасте 13-15 лет.
Диагностика. МРТ головного мозга, как правило, не выявляет никаких отклонений. Но функциональная нейровизуализация показывает изменения в височной доле [14]. При проведении объемного МРТ анализа выявляется уменьшение объема верхней височной извилины [15], в которой локализована речевая зона.
ЭЭГ характеризуются в основном фокусами комплексов острая-медленная волна в задневисочных отведениях, которые часто могут быть мультифокальными и билатерально синхронизированными, существенно усиливающимися при NREM сне. CSWS возникает на определенной стадии заболевания почти во всех случаях, но это не является главным требованием при постановке диагноза, т.к. может сохраняться или исчезать в течение REM сна.
Дифференциальная диагностика. Многие случаи синдрома Ландау-Клеффнера ошибочно принимают за глухоту, аутизм или другие психиатрические заболевания.
Возникновение острой или подострой прогрессирующей афазии у детей 2-8 лет без приобретенного пареза или симптомов энцефалита наиболее вероятно связано с синдромом Ландау-Клеффнера.
Синдром Ландау-Клеффнера также трудно отличить от ECSWS из-за схожих клинических признаков и данных ЭЭГ. Однако главным отличием являются выраженные речевые нарушения при синдроме Ландау-Клеффнера, в то время как при ECSWS главным критерием для постановки диагноза является наличие CSWS на ЭЭГ [16].
Прогноз. Эпилептические приступы, изменения на ЭЭГ исчезают к 15 годам. К этому возрасту также значительно уменьшаются речевые и нейропсихологические расстройства и половина всех больных могут вести относительно нормальную жизнь. Около 10-20% пациентов полностью выздоравливают.
Существует прямая связь между продолжительностью CSWS и степенью нарушения речевых функций [17]. Ранний дебют заболевания связан с плохим прогнозом для речевых функций.
Сноска 4. Ранний дебют синдрома Ландау-Клеффнера коррелирует с плохим прогнозом для речевых функций в дальнейшем.
Очень редко встречаются спонтанные ремиссии через несколько недель или месяцев после начала заболевания.
Лечение синдромов с ESES
Эпилептические приступы при ECSWS и синдроме Ландау-Клеффнера не частые, ограничены возрастом и легко контролируются назначением антиконвульсантов. Однако медикаментозное лечение направлено в первую очередь на уменьшение эпилептиформной активности на ЭЭГ, что уменьшает речевые, поведенческие и когнитивные расстройства.
Единого протокола лечения синдромов с CSWS не разработано. Все традиционные антиконвульсанты не дают удовлетворительного результата. Препараты, предназначенные для лечения фокальных эпилепсий (фенитоин, карбамазепин, вигабатрин) противопоказаны, т.к. могут вызвать значительное усиление эпилептиформной активности и, соответственно, усугубить нейропсихологический дефицит.
Лечение синдромов с CSWS начинается с подбора противоэпилептической терапии. Используются антиконвульсанты широкого спектра действия (вальпроаты: вальпроат натрия (депакин), вальпроевая кислота (конвулекс), леветирацетам (кеппра)) как в монотерапии, так и в сочетании с этосуксимидом (суксилепом), клоназепамом или клобазамом (фризиумом) [5,12,18]. Сультиам (осполот) является препаратом выбора. Клобазам и сультиам на сегодняшний день в России не зарегистрированы.
Были предложены следующие схемы лечения: пероральный прием бензодиазепинов (диазепама, клобазама, клоназепама) в сочетании с вальпроатом короткими циклами (3-4 нед.) [5,12,18]. При применении ректального диазепама (1 мг/кг) наблюдается улучшение [18].
Если лечение антиконвульсантами не дает эффекта, рекомендован АКТГ (тетракозактид (синактен-депо) по 2 мл ежедневно или с интервалами до 3 дней и постепенным уменьшением дозы в течение 3 мес.) или большие дозы кортикостероидов (преднизолон 2-5 мг/кг ежедневно с постепенным уменьшением дозы в течение 3 мес.) [5], особенно у новых пациентов [12]. Продолжительность лечения после этого периода зависит от результатов и побочных эффектов. АКТГ и стероиды применяются вместе с вальпроатами и бензодиазепинами, прием которых продолжается после отмены гормональной терапии.
В некоторых случаях успешно применяется внутривенный иммуноглобулин.
Эффективность лечения оценивается как клинически, так и при помощи повторной регистрации ЭЭГ сна. В случае рецидива CSWS после ранее успешно проведенного курса гормональной терапии целесообразно его повторить.
В резистентных случаях синдрома Ландау-Клеффнера успешно применяется субпиальная интракраниальная транссекция. Эта хирургическая техника позволяет убирать способность кортикальной ткани распространять эпилептиформную активность и генерировать разряды, сохраняя нормальные кортикальные физиологические функции.
Источник: журнал "Медицинский совет" №3-4 2008.
Читайте также:
