
Антеколлис при болезни паркинсона
Н.В. Федорова, О.А. Орехова
Российская медицинская академия последипломного образования, Центр экстрапирамидных заболеваний (Москва)
Кроме того, КК часто сопровождается болевым синдромом. Данные о распространённости и частоте КК при БП противоречивы: от 3,5% (Wenhui Fan et al., 2009) до 27,3% (Weintraub. et al., 2006).
КК при БП обычно появляется по мере прогрессирования заболевания. По данным некоторых авторов, пациенты с БП и КК характеризуются более старшим возрастом, большей длительностью заболевания, более выраженной степенью тяжести БП, большей продолжительностью терапии препаратами леводопы, большей суточной дозой леводопы, ранним присоединением аксиальных симптомов и более частой сопутствующей деменцией. Другие авторы, напротив, не находят корреляции между степенью сгибания туловища и возрастом, длительностью БП, стадиями заболевания и продолжительностью лечения и дозами леводопы. Для клинической оценки степени выраженности КК используют гониометрическое измерение степени тораколюмбальной флексии, рассчитываемой как угол между вертикальной плоскостью и плоскостью, проходящей через край акромиона. Как правило, КК считают переднюю флексию, превышающую угол наклона 45°.
Для дифференциальной диагностики КК из лабораторно-инструментальных тестов важны определение СОЭ, С-реактивного белка, электролитов (кальций и фосфор), креатинфосфокиназы (КФК), витамина Д, пирувата и лактата для исключения миозитов и митохондриальной патологии. Данные лабораторных обследований при КК обычно показывают нормальный или повышенный уровень КФК. По данным ЭМГ поясничных и грудных паравертебральных мышц отмечаются обильные фибрилляции, положительные острые волны и патологические разряды высокой частоты.
При КК целесообразно проведение нейровизуализационных методов исследования (КТ и МРТ грудного и поясничного отделов позвоночника, паравертебральных мышц, головного мозга), позволяющих исключить скелетно-мышечные заболевания или структурные изменения базальных ганглиев, однако данные этих методов при БП с КК не обладают специфичностью. Гистологическое исследование паравертебральных мышц при КК выявляет обширный фиброз и наличие жировой ткани с дегенеративными волокнами. Возможности терапии КК при БП достаточно ограничены. При всей сложности терапии КК в ее основе всегда лежит лечение основного заболевания.
Лечение должно включать консервативные мероприятия, отмену медикаментов, способных вызвать КК, хирургическую коррекцию или глубокую стимуляцию мозга.
В настоящее время определено, что эффективная терапия для КК при БП не найдена. В большинстве описанных в литературе наблюдений КК оказывается нечувствительной к стандартной противопаркинсонической терапии. Только в редких случаях возможно некоторое уменьшение выраженности КК, или же, напротив, ее усиление, чаще при приеме АДР. Поэтому при возникновении КК, связанной с изменениями дофаминергической
Болезнь Паркинсона и расстройства движений
терапии, целесообразна попытка коррекции медикаментозного лечения, прежде всего, исключение или замена одного препарата АДР на другой. Стероиды, по данным различных авторов, являются эффективными лишь в редких случаях БП с КК [26].
Данные об эффективности противосудорожных препаратов при КК противоречивы. Назначение препаратов других фармакологических групп (миорелаксантов, антиконвульсантов) чаще всего не эффективно. Однако, по мнению некоторых авторов, применение противосудорожного препарата клоназепама снижает выраженность КК при БП.
В комплексной терапии при КК обязательным должно быть включение физиотерапевтических и психотерапевтических методик. Иногда аномальное положение туловища улучшается при использовании заплечного рюкзака. Некоторые авторы предлагают использование для облегчения КК классических поясничных корсетов, кожаных ортезов. В настоящее время предлагается использование новых специальных ортезов, изготовленных по принципу торако-тазовой передней фиксации, которые дают неплохие результаты в лечении КК.
Локальные инъекции ботулинистического токсина (БТ) в клинически заинтересованные мышцы (подвздошную мышцу, прямую мышцу живота) при КК, ассоциированной с БП, дают неоднозначные результаты. При этом часто необходимо применение высоких доз БТ, обусловливающих развитие побочных эффектов. Различные авторы отмечают как положительную динамику, так и практически полное отсутствие эффекта на фоне регулярных инъекций БТ.
Хирургическое лечение КК складывается из стабилизирующих вертебральных вмешательств, а также хронической стимуляции глубоких структур мозга (DBS). Среди спектра хирургических операций на позвоночнике применяют заднюю тораколюмбальную фиксацию в сочетании с передним межпозвонковым артродезом на уровне нескольких позвоночных сегментов. При этом почти всегда лечение сопряжено с необходимостью длительной иммобилизации, стационарного наблюдения, повторными хирургическими вмешательствами и связанным с ними высоким риском осложнений.
В современной литературе постепенно накапливаются данные об использовании DBS в лечении ассоциированной с БП КК. Однако результаты применения DBS в этих случаях остаются противоречивыми. Кроме того, среди исследователей не сложилось единого мнения относительно выбора оптимальной структуры для DBS.
Таким образом, КК встречается при многих неврологических заболеваниях, самым частым из которых является БП. КК проявляется патологической позой с насильственным наклоном туловища вперед. Она возникает в вертикальном положении, усиливается при длительном стоянии или ходьбе и уменьшается в горизонтальном положении. В качестве причин развития КК при БП предполагают сегментарную аксиальную дистонию или локальную миопатию паравертебральных мышц, однако точные механизмы патогенеза изучены недостаточно. КК, ассоциированная с БП, значительно инвалидизирует пациентов, приводит к нарушению передвижения и самообслуживания.
Цель исследования: Определить клинические проявления синдрома КК при БП и влияние его на качество жизни больных.
Пациенты и методы исследования
В исследование было включено 90 больных БП, разделенных на две группы. Основную группу составили 70 больных БП с КК, средний возраст пациентов составил 68,9 ± 7,8 лет (от 53 до 86). Группу сравнения составили 20 пациентов БП без КК. Группа сравнения соответствовала основной по возрасту, полу, продолжительности заболевания и степени тяжести. Средняя степень тяжести по шкале Хен–Яру составила 3,4 ± 0,7 баллов, средняя продолжительность БП – 9,9 ± 5,6 лет. У 46 больных отмечалась смешанная форма заболевания (66%), у 19 – акинетико-ригидная форма (27%), дрожательно-ригидная форма наблюдалась у 5 больных (7%). Для оценки степени тяжести БП использовалась шкала Хен–Яра в модификации Линдвалла (Hoehn, Jahr, 1967; Lindvall et al., 1987); для оценки выраженности основных симптомов БП – унифицированная рейтинговая шкала БП (Unifed Parkinsons Disease Rating Scale – UPDRS, Fahn. et al., 1987); для оценки постуральной нестабильности – шкала Берга (Berg et al., 1992), шкала Тиннети (Tinetti et al., 1986), шкала нарушений ходьбы и равновесия (Gait and Balanse Scale, Jancovic, 2002); для оценки повседневной активности больных – шкала Schwab и England (1967); для оценки качества жизни больных БП – шкала PDQ-39 (Peto et al., 1995); для оценки степени выраженности КК и степени выраженности болевого синдрома – опросник КК (Margraf et al., 2010); для оценки влияния дофаминергических препаратов на выраженность КК использовался дневник Хаузера (Hauser et al., 2004).
Результаты и обсуждение
Средняя длительность КК составила 3,6 ± 3,0 лет. В общей группе преобладали пациенты с возрастом дебюта БП от 51 до 60 лет, в то время как КК возникала преимущественно у пациентов в возрасте от 61 до 70 лет. У 55 больных (78,6%) отмечался наклон вперед, у 15 больных (21,4%) – сочетание наклона вперед с наклоном в стороны. Тораколюмбальный угол составил 63,5±17,2 градусов, цервикокраниальный угол – 43,2±22,5 градусов. Болевой синдром наблюдался у 62 больных (88,6%). У 53 больных (75,7%) отмечалась четкая связь между появлением болевого синдрома в позвоночнике и началом развития КК. Болевой синдром был представлен в виде корешковой симптоматики у 20 пациентов (32,2%), в виде мышечно-тонического синдрома – у 37 пациентов (59,7%), у 5 пациентов (8,1%) наблюдалось сочетание корешкового и мышечно-тонического синдромов. Выраженность болевого синдрома оценивалась по опроснику КК и составила 54,9 ± 24,5%. Предшествующий вертеброгенный анамнез был выявлен у 46 больных (66%) в виде компрессионных переломов тел поясничных позвонков на фоне остеопороза, протрузий и грыж дисков на пояснично-крестцовом уровне, подтвержденных на основании КТ или МРТ пояснично-крестцового отдела позвоночника; имелись также проявления остеопороза, остеохондроза, спондилоартроза, подтвержденные данными рентгенографии грудного и пояснично-крестцового отделов. У 44 пациентов КК возникала уже в положении сидя (62,9%), у 21 – сразу при приеме вертикального положения (30%), у 2 – после прохождения 50 м (2,9%), у 3 (4,3%) – только после значительной нагрузки в вертикальном положении. У 54 пациентов (77,1%) КК наблюдалась в течение всего дня, у 4 (5,7%) – только в течение нескольких часов в дневное время, у 12 (17,1%) – возникала только вечером. КК вызывала ограничение повседневной активности на 51,6±25,6%. Факторами, усиливающими проявления КК, были статические нагрузки (2,9%), в 10% – стрессовые ситуации; физические нагрузки провоцировали развитие КК у 40 больных (57,1%); стрессовые и физические нагрузки вместе вызывали ухудшение КК у 6 больных (8,6%).
Для коррекции КК 10 пациентов (14,3%) использовали корсет; одному пациенту (1,4%) помогало ношение заплечного рюкзака; применение рюкзака и корсета были эффективны в одном случае (1,4%), в то время как 58 пациентов (82,9%) не использовали корригирующие приемы для уменьшения выраженности КК. Тридцать шесть пациентов (37,1%) с КК использовали при ходьбе трость, 4 (5,7%) были вынуждены использовать ходунки; сочетание трости и ходунков использовали 4 пациента (5,7%), 26 пациентов (37%) не нуждались в помощи при ходьбе.
Падения наблюдались у 29 пациентов (41,4%).
У большинства больных (65,7%) преобладал медленно прогрессирующий тип течения КК – постепенное начало в течение 1–6 месяцев и нарастание КК на протяжении нескольких лет.
Общая сумма баллов по шкале повседневной активности Schwab и England составила 68,3±19,5 баллов в основной группе и 72,0±15,8 баллов в группе сравнения; по шкале выраженности основных симптомов БП (UPDRS) – 54,7 ±17,9 баллов в основной группе и 53,2±18,9 баллов в группе сравнения; по шкале оценки качества жизни больных БП PDQ-39 – 106,0±21,3 баллов в основной группе и 100,1±25,3 баллов в группе сравнения).
У 58 больных (82,9%) четкой связи между появлением КК и началом приема дофаминергических препаратов не отмечалось, только у 18 больных (25,7%) выявлена четкая связь появления КК с началом дофаминергической терапии.
У 27 больных (38,6%) отмечалось ухудшение позы в off-периоде, у 8 пациентов (11,4%) в утренние часы после ночного перерыва в приеме препаратов отмечалось явное ухудшение позы, нивелирующееся в дневное время. Тридцати пяти пациентам с БП, у которых отмечалось нарастание позных расстройств в период выключения и в утренние часы (дистонии периода выключения и раннего утра), для удлинения эффекта однократной дозы леводопы и стабилизации концентрации препарата в крови был назначен комбинированный препарат препарат леводопа/карбидопа/энтакапон (Сталево) в средней суточной дозе 610 мг. Применение такого препарата, включающего ингибитор фермента катехол-О-метилтрансферазы (КОМТ) – энтакапон, является одним из конструктивных подходов к улучшению эффективности леводопатерапии. Процесс метилирования леводопы происходит на периферии (в желудочно-кишечном тракте и в кровеносном русле) и в головном мозге. При этом КОМТ отвечает за 10% катаболизма леводопы на периферии. Метилирование может быть заторможено путем назначения ингибитора КОМТ периферического действия энтакапона (рис. 1).

Рисунок 1. Оптимизация фармакокинетики леводопы.
Ингибиторы КОМТ увеличивают биодоступность леводопы, уменьшая уровень ее неактивных метаболитов. Энтакапон не проходит ГЭБ и препятствует метилированию леводопы в желудочно-кишечном тракте и кровеносном русле, благодаря этому сохраняется более высокий уровень леводопы, которая после прохождения ГЭБ служит материалом для синтеза дофамина в головном мозге. На фоне приема энтакапона период полужизни леводопы удлиняется на 25–75% (рис. 2).

Рисунок 2. Изменение периода полужизни леводопы в плазме при добавлении энтакапона.
Заключение. Таким образом, согласно клиническим наблюдениям, большинство пациентов страдали от КК даже в положении сидя (62,9%), КК наблюдалась в течение всего дня (77,1%), ухудшение КК отмечалось после физической нагрузки или стресса (57,1% и 10% соответственно). В основном пациенты использовали для передвижения трость (37,1%); некоторые из-за тяжести КК даже вынуждены были использовать ходунки (5,7%). Более отдаленным последствием КК являлись нарастание постуральной нестабильности и увеличение частоты падений. У большинства пациентов БП с КК наблюдались боли в спине (88,6%) и имелся предшествующий вертеброгенный анамнез (66%). В значительном большинстве случаев КК была более инвалидизирующим фактором для пациентов, чем классические двигательные симптомы БП. У 25,7% пациентов выявлена четкая связь между появлением КК и началом дофаминергической терапии. У 51,4% больных отмечалась четкая зависимость степени выраженности КК от моторных флюктуаций: ухудшение в период выключения действия дофаминергических препаратов зафиксировано у 38,6% больных; у 11,4% пациентов отмечалось ухудшение позы в утренние часы. В данном исследовании выявлена эффективность нового трехкомпонентного препарата леводопа/карбидопа/энтакапон (Сталево) в лечении позных расстройств (дистонии мышц туловища) периода окончания дозы за счет стабилизации концентрации леводопы в крови и удлинения периода ее полужизни в плазме.
"Мой супруг — офицер, человек очень уравновешенный. Он всю жизнь работал, ни на что не жаловался, я никогда не слышала от него про усталость. Но приблизительно шесть лет назад он стал слишком тихим, мало разговаривал — просто сидел и смотрел в одну точку. Мне даже в голову не пришло, что он болен. Наоборот, ругала, что раньше времени постарел. Примерно тогда же к нам приехала двоюродная сестра из Англии — она работает в больнице — и сразу сказала, что у Рафика все очень плохо, нужно завтра же вести его к врачу. Так мы узнали о болезни Паркинсона", — вспоминает Седа из Еревана.
Что такое болезнь Паркинсона
Паркинсон — одна из самых страшных фамилий, что можно услышать в кабинете у невролога. Ее носил английский врач, который в 1817 году подробно описал шесть случаев загадочной болезни. День рождения Джеймса Паркинсона, 11 апреля, и выбран памятной датой Всемирной организацией здравоохранения. Из-за основных симптомов Паркинсон называл недуг дрожательным параличом: движения больных замедляются, становятся скованными, мышцы сильно напрягаются, а руки, ноги, подбородок или все тело бесконтрольно трясутся. Впрочем, в четверти случаев дрожания — самого известного признака болезни — нет.
Все это напоминает обыкновенную старость. Двигательные симптомы — собирательно их называют паркинсонизмом — встречаются у многих здоровых стариков. Но болезнь Паркинсона этим не исчерпывается. На поздних стадиях человек легко теряет равновесие, то и дело застывает на месте во время ходьбы, ему трудно говорить, глотать, спать, появляются тревога, депрессия и апатия, мучают запоры, падает кровяное давление, слабеет память, а под конец часто развивается слабоумие. Самое печальное — вылечить болезнь Паркинсона пока невозможно.
В начале XX века российский невропатолог Константин Третьяков выяснил, что при болезни Паркинсона гибнут клетки черной субстанции, области мозга, которая частично отвечает за движения, мотивацию, обучение. Что вызывает смерть нейронов, неизвестно. Возможно, дело в сбоях внутри клеток, но еще замечено, что внутри них скапливается вредный белок. Оба процесса наверняка как-то связаны, но ученые не знают, как именно.
В 2013 году физиолог Сьюзан Гринфилд из Оксфордского университета представила новую модель развития нейродегенеративных заболеваний, в том числе болезней Паркинсона и Альцгеймера. Гринфилд предположила, что при повреждении мозга, например, от сильного удара выделяется особое вещество. У маленьких детей из-за него растут новые клетки, а на взрослых оно, судя всему, действует противоположным образом, дальше повреждая клетки. После этого следует еще больший выброс вещества, и цепная реакция постепенно разрушает мозг. По злой иронии взрослые впадают в младенчество из-за фермента, необходимого младенцам.

Впрочем, догадка Гринфилд объясняет не все. Болезнь Паркинсона связана с наследственностью: близкий родственник с таким же диагнозом или тремором другой природы — главный фактор риска. На втором месте — запоры: иногда их вызывают изменения в мозге, когда еще не появились двигательные симптомы. Также риск растет, если человек никогда не курил, живет за городом, пьет колодезную воду, но при этом сталкивался с пестицидами, а снижается — у любителей кофе, алкоголя и гипертоников. В чем тут секрет, непонятно, как непонятно, почему болезнь Паркинсона обычно начинается в старости: если на пятом десятке лет болеет примерно один из 2500 человек, то на девятом — уже один из 53.
Новую зацепку дала свежая работа ученых из Университета Томаса Джефферсона: возможно, болезнь Паркинсона связана с иммунной системой. Исследователи взяли мышей с мутантным геном, который часто встречается у больных, и ввели им безвредные остатки бактерий. Из-за этого у зверьков началось воспаление, затронувшее и мозг, причем иммунных клеток было в 3–5 раз больше, чем у обычных мышей. Из-за этого в мозге мутантов начались процессы, губительные для нейронов черной субстанции. Как и в модели Гринфилд, процессы эти оказались циклическими: воспаление в мозге может остаться даже после того, как тело справилось с инфекцией. Впрочем, сами авторы исследования признаются, что в этом механизме еще многое не ясно.
Каково живется больным и их близким
В России болезнь Паркинсона есть примерно у 210–220 тыс. человек. Но эти данные рассчитаны по косвенным показателям, а единого реестра не существует. Анастасия Обухова, кандидат медицинских наук с кафедры нервных болезней Сеченовского университета и специалист по болезни Паркинсона, считает эту статистику заниженной. "Многие больные впервые приходят уже на развернутых стадиях болезни. При расспросе удается выяснить, что признаки появились еще несколько лет назад. У большинства наших людей действует принцип "Пока гром не грянет, мужик не перекрестится": они читают в интернете, спрашивают соседок, а к врачу не обращаются. Это в Москве, а в маленьких городках и поселках к врачу идут только если совсем помирают", — объясняет Обухова.
Вдобавок попасть на прием не так-то просто. Для этого сначала нужно сходить к терапевту, чтобы тот направил к неврологу. Но и тогда нет гарантии, что человеку поставят правильный диагноз и назначат нужное лечение. "Врач в поликлинике не может разбираться во всем, поэтому должен послать больного к узкому специалисту. А окружных паркинсонологов, по-моему, убрали. Во всяком случае, пациенты на это жаловались", — рассказывает Обухова. Правда, если больной все-таки попал к нужному доктору, лечить его будут на мировом уровне. Оттого в Россию с болезнью Паркинсона прилетают даже из других стран.
Одиссею по кабинетам приходится часто повторять, потому что болезнь прогрессирует — терапию нужно подстраивать. Лечение обходится дорого: месячный запас некоторых лекарств стоит по 3–5 тыс. рублей, а на поздних стадиях назначают сразу несколько препаратов. "В районных поликлиниках лекарства иногда дают бесплатно, но только дешевые дженерики. Комментировать их качество не буду. Иногда нужных лекарств нет. Тогда их заменяют чем-то другим. Пациентам от этого плохо", — объясняет Обухова.
*Импакт фактор за 2018 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Читайте в новом номере
ММА имени И.М. Сеченова
Клиническая картина

Болезнь Паркинсона. На рисунке А показан участок мозга здорового человека с хорошо пигментированным черным веществом. На рисунке В - участок мозга пациента, страдающего болезнью Паркинсона. Заметно отсутствие пигментации черного вещества.
П – это синдром, который может встречаться в рамках разных заболеваний. Наиболее часто причиной П является идиопатический паркинсонизм, или болезнь Паркинсона (БП). П часто наблюдается в рамках других идиопатических дегенеративных заболеваний нервной системы. Последние часто называют паркинсонизмом–плюс. К этой группе относятся мультисистемная атрофия, прогрессирующий надъядерный паралич, болезнь диффузных телец Леви, кортико-базальная дегенерация. Нередко встречается симптоматический П (не связанный с первичным дегенеративным заболеванием нервной системы). К нему относят лекарственный, сосудистый, посттравматический, постэнцефалитический, токсический П, П при опухолях головного мозга и гидроцефалии. П характеризует также клиническую картину некоторых наследственных дегенеративных заболеваний ЦНС. К их числу относятся гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова), спиноцеребеллярные атаксии, семейная кальцификация базальных ганглиев, болезнь Гентингтона и др. Таким образом, синдром П не является синонимом БП.
Диагноз и дифференциальный диагноз
Эссенциальный тремор
На первом этапе диагностической работы необходимо убедиться, что у больного действительно имеется синдром П. Наиболее часто за истинный П ошибочно принимают эссенциальный тремор (ЭТ) и своеобразные изменения походки, связанные с сосудистой патологией головного мозга. Таким пациентам очень часто неправильно ставят диагноз БП.
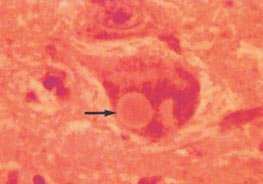
Тельца Леви при болезни Паркинсона. В цитоплазме нейрона определяется эозинофильное ядро окруженное неокрашенной зоной.
Последние, как правило, выявляют множественные мелкие очаги сосудистого происхождения (лакуны) в области базальных ганглиев и/или изменения белого вещества полушарий головного мозга (лейкоареоз). Следует иметь в виду, что схожая клиническая картина может возникать иногда при нормотензивной гидроцефалии и реже – при опухолевом поражении головного мозга. При нормотензивной гидроцефалии наряду с нарушением ходьбы по типу описанного выше имеют место деменция и расстройство функции тазовых органов (эту триаду симптомов называют триадой Хакима–Адамса). МРТ головы, как правило, обнаруживает резко расширенные боковые желудочки.
Болезнь Паркинсона
Диагноз БП – это клинический диагноз. Параклинические методы исследования обычно выявляют неспецифические изменения. Нередко у больных с классической БП в головном мозге имеются сосудистые изменения. Это, однако, ни в коем случае не является свидетельством сосудистого генеза болезни. Просто у большинства пожилых людей, в том числе практически здоровых, подобные изменения могут иметь место. Тем не менее, в некоторых случаях на БП наслаивается атеросклеротический псевдопаркинсонизм, что может быть причиной необычной для БП походки и ранних постуральных нарушений.
Лекарственный паркинсонизм
Лекарственный П может быть обусловлен препаратами, которые воздействуют на пресинаптические дофаминовые нейроны черной субстанции, истощая запасы дофамина в них (например, резерпин) или, наиболее часто, нейролептиками, которые блокируют постсинаптические дофаминовые рецепторы, такими как производные фенотиазина (хлорпромазин), бутирофеноны (галоперидол), тиоксантины (флупентиксол) и бензамиды (сульпирид). Эти препараты часто применяются при психических заболеваниях. Также П может быть вызван прохлорперазином (используется при рвоте, головокружении и неустойчивости), метоклопрамидом (применяется при заболеваниях желудочно-кишечного тракта, для купирования тошноты и рвоты). П может быть обусловлен также циннаризином, который является атипичным блокатором кальциевых каналов (применяется при вестибулярных расстройствах). Комбинация нейролептиков и антидепрессантов также может быть причиной П.
В психиатрических лечебницах лекарственный П встречается часто. Гипомимия и ахейрокинез – настолько обычное явление среди этого контингента больных, что психиатры часто даже не обращают на них внимание. Тремор встречается менее часто, но может иметь вид классического паркинсонического дрожания. Более того, лекарственный П может быть асимметричным, подобно БП, и нередко совершенно не отличим от БП. Одним из признаков, по которому следует заподозрить лекарственный П, является наличие наряду с акинетико-ригидным синдромом насильственных движений в виде, например, оро-мандибулярной дискинезии (непроизвольные жевательные и/или сосательные движения) или дистонических явлений (спастической кривошеи, окулогирных кризов), стереотипий, акатизии (неусидчивость). Нередко тяжелый лекарственный П сопровождается выраженной дизартрией и дисфагией. Индивидуальная чувствительность к дофаминблокирующим препаратам очень разнообразна. Некоторые пациенты без каких-либо проблем переносят длительное лечение большими дозами этих веществ, тогда как у других побочные явления развиваются уже на малых дозах. Чаще, однако, признаки лекарственного П возникают при приеме больших доз нейролептиков. Лекарственный П обычно развивается постепенно в течение дней или недель. У большинства больных первые признаки появляются через 3 нед после начала лечения. Наиболее часто встречающиеся дебютные признаки – гипомимия и недостаточное раскачивание рук во время ходьбы.
Течение лекарственного П может быть различным. В большинстве случаев он постепенно, в течение нескольких недель, а иногда и дней, проходит после прекращения приема вызвавшего его препарата. Тем не менее нередки случаи, когда П длится в течение месяцев, иногда почти год. Такая ситуация наблюдается при применении нейролептических препаратов, способных к депонированию. В редких случаях лекарственный П не проходит и продолжает прогрессировать, несмотря на прекращение приема вызвавшего его агента. Подобные случаи чаще встречаются среди пожилых людей. Считается, что в таких случаях прогрессирует не сам по себе лекарственный П, а начинает развиваться БП.
Мультисистемная атрофия
Мультисистемная атрофия (МСА) – это спорадическое заболевание, возникающее у взрослых лиц, при котором в отличие от БП дегенерации подвергается не только нигро-стриарная система, но также множество других образований ЦНС, включая мозжечок и его связи, пирамидные пути и образования вегетативной нервной системы (отсюда и происходит название болезни). Соответственно клинически МСА характеризуется сочетанием П, мозжечковых нарушений, пирамидных расстройств и прогрессирующей вегетативной недостаточности (ПВН). П при МСА обусловлен не только поражением клеток черной субстанции, что вызывает дефицит дофамина, но также дегенерацией тех постсинаптических рецепторов, с которыми должен взаимодействовать дофамин.
В клинической картине МСА может преобладать та или иная симптоматика. Те случаи, при которых на первый план выступает П, обозначают термином нигро-стриальная дегенерация (СНД); если в клинической картине ведущим является мозжечковый синдром, это состояние называют оливо-понто-церебеллярной атрофией (ОПЦА); случаи, когда ядром клинической картины является ПВН, обозначают эпонимическим названием – синдром Шая–Дрейджера (СШД).
Несмотря на все различия, нередки ситуации, когда МСА невозможно отличить от БП. В таких случаях следует в основном ориентироваться на эффект препаратов леводопы. При БП эти препараты оказывают драматический положительный эффект, тогда как при МСА этот эффект не столь выражен, кратковременен, а нередко отсутствует совсем. Это обусловлено поражением постсинаптических рецепторов, с которыми должна взаимодействовать леводопа.
Признаки, при наличии которых у больных с П следует подумать об МСА, следующие: быстрое прогрессирование симптоматики; рано развившиеся нарушения равновесия и падения; отсутствие улучшения при лечении препаратами леводопы или недостаточный эффект этих препаратов; ПВН; пирамидные и/или мозжечковые знаки; похолодание, зябкость конечностей; контрактуры; диспропорциональный антеколлис; выраженная дисфония, дисфагия или дизартрия; респираторный стридор; нерегулярный тремор или миоклонии.
Прогрессирующий надъядерный паралич
Болезнь диффузных телец Леви
В последнее время стали выделять новую нозологическую форму, протекающую с синдромом П – болезнь диффузных телец Леви (БДТЛ). Тельца Леви – это внутриклеточные эозинофильные цитоплазматические включения, которые обнаруживаются в клетках черного вещества при БП и считаются маркером этой болезни. При БДТЛ они встречаются не только в черном веществе, но в большом количестве широко диссеминированы по всему головному мозгу. Диагноз БДТЛ – патоморфологический диагноз. Клинически это заболевание характеризуется П, который обычно хорошо лечится препаратами леводопы, наряду с деменцией с выраженными зрительными галлюцинациями. Типичным является также флуктуация выраженности расстройств высших психических функций – в основном за счет изменения способности концентрации внимания.
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) и другие заболевания
Существует несколько более редких причин истинного или псевдопаркинсонизма. Одна из них, о которой следует всегда вспомнить при наличии П у людей моложе 45 лет (в том числе у детей), это гепатолентикулярная дегенерация, или болезнь Вильсона-Коновалова. Это наследственное заболевание, при котором отмечается нарушение метаболизма меди в организме из-за недостаточности фермента церулоплазмина. В результате медь в избыточном количестве откладывается в печени, базальных ганглиях и вокруг радужной оболочки глаза. Болезнь Вильсона-Коновалова следует подозревать не только при наличии П у молодых людей, но и при возникновении у них других признаков поражения экстрапирамидной системы (например, дистонии) или психических расстройств. Диагностика основана на обнаружении с помощью щелевой лампы отложения меди вокруг радужки – кольцо Кайзера–Флейшера. Последнее на стадии неврологических проявлений имеет место у 98% больных. Диагностическое значение имеет также исследование экскреции меди с мочой и концентрации церулоплазмина в крови. Последнее, однако, в отсутствие кольца Кайзера–Флейшера и нормальной экскреции меди не имеет диагностической значимости. Если ситуация продолжает оставаться неясной, проводится биопсия печени или генетическое тестирование. Болезнь Вильсона-Коновалова довольно успешно лечится с помощью D-пеницилламина и препаратов цинка в сочетании с диетой.
Токсический П может иметь место при интоксикации марганцем. Нередко встречается П, вызванный токсином МФТП (1-метил-4-фенил- 1, 2, 3, 6-тетрагидропиридин), так как оно избирательно поражает черную субстанцию, вызывая гибель дофаминовых нейронов. Схожий с этим веществом токсин вырабатывается при кустарном приготовлении некоторых наркотических веществ, поэтому П нередко наблюдается среди молодых людей, страдающих наркоманией. Наряду с П при этом могут отмечаться пирамидные и псевдобульбарные симптомы.
П встречается при энцефалопатии боксеров. Считается, что в таком случае этиологическую роль играют регулярно получаемые множественные черепно-мозговые травмы. Следует помнить, что энцефалопатия боксеров – практически единственная ситуация, когда черепно-мозговые травмы играют роль в развитии П. В целом у людей, не занимающихся боксом, наличию черепно-мозговой трамы в анамнезе (одной или нескольких) не придается этиологическое значение.
П иногда может иметь место при хорее Гентингтона, которое обычно протекает с выраженными гиперкинезами. Это наследственное заболевание обычно дебютирует на четвертом десятилетии жизни, однако встречаются случаи и более раннего начала. Обычно с акинетико-ригидным синдромом протекает так называемая ювенильная форма хореи Гентингтона (форма Вестфаля). Встречаются также семейные случаи П, в том числе ювенильного. Поэтому у всех больных, имеющих синдром П, сбор наследственного анамнеза имеет большое значение.
Акинезия и ригидность могут иногда наблюдаться при болезни Альцгеймера, Пика, при которых ядром клинической картины является прогрессирующая деменция. При болезни Альцгеймера в первую очередь страдает память, а при болезни Пика на первый план выступает распад личности. При этих заболеваниях П чаще проявляется умеренной акинезией, ригидность выражена меньше, а паркинсонический тремор практически никогда не наблюдается.
Постэнцефалитический П в наше время практически не встречается. Много случаев наблюдалось после пандемии летаргического энцефалита в начале XX столетия. С тех пор в литературе описано только несколько случаев.
Особая эндемическая форма П наблюдается на острове Гуам и в некоторых других местах восточной части Тихого океана. В этих местах отмечается много случаев П в сочетании с амиотрофиями и деменцией (гуамский комплекс паркинсонизм – деменция). Патоморфологически мозг этих больных выглядит, как мозг больных ПНП. Одинаковыми являются также гистологические маркеры.
Читайте также:
