
Астроциты альцгеймера ii типа
Подтверждена разрушительная роль активации астроцитов при болезни Альцгеймера
Группа ученых из Университета Кентукки во главе с адъюнкт-профессором кафедры молекулярной и биохимической фармакологии Медицинского колледжа UK Кристофером Норрисом (Christopher Norris), PhD, опубликовала статью, впервые доказательно подтверждающую пагубную роль активации астроцитов в развитии болезни Альцгеймера (Furman et al., Targeting Astrocytes Ameliorates Neurologic Changes in a Mouse Model of Alzheimer's Disease). Кроме того, Центр по проблемам старения Сэндерса-Брауна (The UK Sanders-Brown Center on Aging), научным сотрудником которого является доктор Норрис, получил значительные средства от Национальных Институтов Здравоохранения (National Institutes of Health) США на дальнейшее развитие этого исследовательского направления.

С помощью генной инженерии, используя в качестве вектора аденоассоциированный вирус (AAV), содержавший специфический для астроцитов промоутер Gfa2, исследователи сделали мишенью своего воздействия астроциты гиппокампа мышей с моделью болезни Альцгеймера (линия APP/PS1) и, таким образом, изменили степень активации этих клеток. Векторы AAV-Gfa2 стимулировали экспрессию VIVIT – пептида, вмешивающегося в сигнальный путь кальциневрина/NFAT (ядерного фактора активированных Т-клеток), ответственного за биохимические каскады, ведущие к активации астроцитов.
Эта генетическая манипуляция была проведена до развития у мышей выраженной амилоидной патологии, и состояние животных было оценено через 10 месяцев по нескольким биомаркерам болезни Альцгеймера. Все это время мыши получали Gfa2-VIVIT.
Ученые пришли к выводу, что подавление активации астроцитов приводит, в свою очередь, к подавлению активации микроглии (клеток-посредников нейровоспаления), снижает уровни токсичного амилоида, положительно влияет на синаптическую пластичность и когнитивные способности животных.
Таким образом, исследование подтверждает разрушительную роль активации астроцитов при болезни Альцгеймера и правильность концепции их терапевтического таргетинга. По мнению доктора Норриса и его коллег, полученные результаты закладывают основу для разработки методов лечения пациентов, страдающих болезнью Альцгеймера, или, возможно, другими нейродегенеративными заболеваниями, мишенью которых будут астроциты.
Читать статьи по темам:
Читать также:
Новая надежда в лечении болезни Альцгеймера
Новый подход, заключающийся в блокировании одной из сигнальных молекул иммунной системы, значительно снижает выраженность изменений, характерных для болезни Альцгеймера.
Клетки хориоидного сплетения помогут в борьбе с болезнью Альцгеймера?
Выращенные из эмбриональных стволовых клеток эпителиальные клетки хориоидного сплетения могут помочь в лечении болезнь Альцгеймера и других нейродегенеративных заболеваний.
Болезнь Альцгеймера: наступление по всем фронтам
Сентябрь 2012 года получил статус первого всемирного месяца болезни Альцгеймера. В связи с этим профессор Филипп Амуэль, директор программы DISTALZ, отвечает на вопросы корреспондента индийского сайта Express Pharma.
Отвергнутый препарат от нейродегенерации частично реабилитирован
Латрепиридин – перспективный препарат для лечения болезни Альцгеймера, отвергнутый после неудачи клинического исследования фазы III, – реабилитирован в результате исследования, вскрывшего два механизма его действия.
Вакцина от болезни Альцгеймера: ищем добровольцев
Медицинский центр Джорджтаунского университета набирает пациентов с начальными стадиями болезни Альцгеймера для проведения клинического исследования вакцины против этого заболевания.
Электронное СМИ зарегистрировано 12.03.2009
Свидетельство о регистрации Эл № ФС 77-35618

В настоящее время болезнь Альцгеймера (БА) является наиболее распространенным, длительно протекающим нейродегенеративным заболеванием. На начальном этапе болезнь проявляется нарастающим ухудшением кратковременной памяти. Позднее наблюдаются поведенческие нарушения, происходит потеря долговременной памяти, затем следует деградация систем жизнеобеспечения организма, что в конечном итоге приводит к смерти [1]. Отложение амилоида β (Aβ) во внеклеточном матриксе головного мозга (ГМ) является типовым патологическим признаком при БА. В основном это Аβ40 и Aβ42 (по количеству аминокислот), причем Aβ42 более склонен к агрегации и более токсичен для клеток мозга [2]. Токсичность Aβ объясняется его биохимическими свойствами, способствующими дисмеризму в так называемые бляшки, воздействующие на нейроны и клетки глии. По данным исследований, амилоидные бляшки вызывают воспалительный ответ [3] и сопутствующую дисфункцию митохондрий [4]. Снижается синаптическая пластичность, происходят сбои во внутриклеточных сигнальных каскадах, в результате нейроны деградируют [5]. Бляшки – не единственный признак, внутри нейронов при БА наблюдаются нейрофибриллярные клубки (NFT) – скопления нитей гиперфосфорилированного Тау-белка [6]. В норме этот белок связан с тубулином микротрубочек и стабилизирует их структуру. Гиперфосфорилированнный Тау диссоциирует от микротрубочек, вызывая коллапс микротрубочкового цитоскелета, в результате нарушаются аксональный транспорт и синаптический метаболизм [7]. Исследования, направленные на ингибирование гликогенсинтетазы киназы-3β (GSK3β), фермента, регулирующего фосфорилирование Тау-белка (сверхактивная GSK3β приводит к гиперфосфорилированию Тау), предотвращают деградацию нейронов [8]. Чрезмерная активность GSK3β на ранних стадиях БА может вызывать перепроизводство Aβ и в дальнейшем гиперфосфориляцию Тау-белка [9]. Однако, несмотря на весомые аргументы за первостепенную роль Aβ, в исследованиях не была обнаружена устойчивая корреляция между плотностью и количеством бляшек в мозге и тяжестью деменции [10]. Корреляция прослеживается с плотностью клубков гиперфосфорилированного Тау, но не объясняет все патологические изменения [10]. Основным же коррелятом ухудшений при БА является потеря синапсов и холинергических нейронов, что и вызывает когнитивные нарушения [11, 12]. В холинергических нейронах снижено производство холин-ацетилтрансферазы (СhAT), катализирующей реакцию образования нейромедиатора ацетилхолина [13]. Ингибиторы ацетилхолинэстеразы (AChE), фермента, катализирующего гидролиз ацетилхолина до холина и уксусной кислоты, повышают уровень ацетилхолина в синаптической щели и частично улучшают состояние пациентов, но на очень непродолжительный период, не прекращая дальнейшую дегенерацию [14]. Несмотря на многочисленные исследования, в основном ориентированные на нейронные процессы, на сегодняшний день не разработано ни одного препарата, позволяющего вылечить болезнь. Комплекс профилактических мер, включающий в себя диету, физическую активность и умственные нагрузки, показывает некоторую эффективность в борьбе с заболеванием [15].
Заболевание классифицируют на раннюю и позднюю формы. Поздняя (спорадическая) форма наиболее распространена (более 90% случаев) среди людей старше 65 лет [1]. Основным генетическим фактором риска для позднего начала заболевания является ген APOE в хромосоме 19 [1, 16]. Генетически наследуемая форма БА с ранним началом (до 60–65 лет) может быть вызвана следующими мутациями: в гене APP, который кодирует белок-предшественник амилоида (БПА), в гене пресенелина 1 (PSEN1) в хромосоме 14 и/или в гене пресенелина 2 (PSEN2) в хромосоме 1 [1, 16]. Мутации в пресенелинах приводят к изменению обработки БПА и, кроме того, к нарушению передачи Ca2+-сигналов. Так, мутации PSEN1 приводят к увеличению выделения Ca2+ из эндоплазматического ретикулума (ЭР) с помощью рецепторов инозитолтрифосфата (IP3) [17]. Не все наследуемые формы БА объясняются мутациями в вышеупомянутых генах [1]. К дополнительным факторам риска БА следует отнести (по нарастающей) сахарный диабет, курение, низкий уровень образования, гипертонию, депрессию, физическую инертность [15].
Роль астроцитов в болезни Альцгеймера
Глиальные клетки в ГМ поддерживают гомеостаз, выполняют трофическую, опорную, барьерную и секреторную функции. Нарушения в работе глиальных клеток могут в значительной степени способствовать прогрессированию нейродегенеративных заболеваний или даже стать их причиной. При БА в ответ на образование амилоидных бляшек астроциты и микроглия продуцируют широкий спектр провоспалительных факторов, таких как интерлейкин-1 (IL-1) и фактор некроза опухолей α (TNFα), влияя на иммунную систему мозга [18]. Переключение астроцитов на новый функциональный профиль, так называемая активация, нарушает выполнение их основных функций и приводит к гибели нейронов [19, 20, 21]. Активированные астроциты частично фагоцитируют бляшки Aβ, но чрезвычайно медленно и неэффективно [22]. Протофибриллы Aβ42 длительное время хранятся в астроцитах, что вызывает внутриклеточные дефекты, еще сильнее снижающие способность астроцитов к деградации амилоида [22]. По мере прогрессирования БА наблюдаются увеличение количества и размера астроцитов, окружающих амилоидные бляшки, и атрофия астроцитов, находящихся на периферии пораженной зоны, – астроглиоз [23]. Этот процесс включает молекулярные и морфологические изменения, приводящие к увеличению сомы астроцита из-за перепроизводства белков цитоскелета, однако его прерывание снижает нейрональную пластичность и регенерацию ЦНС [21, 24, 25]. При БА также ухудшается способность астроцитов к поддержанию гематоэнцефалического барьера (ГЭБ) и к поглощению глутамата из синаптической щели (рис. 1) в трехстороннем синапсе [26, 27]. Кроме того, исследования, проведенные на трансгенных мышах с БА, показали, что астроциты демонстрируют увеличение концентрации внутриклеточного цитоплазматического кальция ([Ca2+]i) и усиление передачи Ca2+-сигналов [28]. Гиперактивные спонтанные волны астроцитарного кальция, независимые от нейрональной активности и наблюдаемые во многих трансгенных моделях мышей, способны влиять на передачу сигналов в синапсах [23, 28]. Синаптическая дисфункция при БА коррелирует с тяжестью когнитивного спада и ростом числа активированных астроцитов [29]. Таким образом, хотя астроцитарная активация играет защитную роль в головном мозге при БА, реактивные астроциты с нарушенной в них передачей Ca2+-сигналов могут усугублять повреждения нейронов и ускорять прогрессирование заболевания.
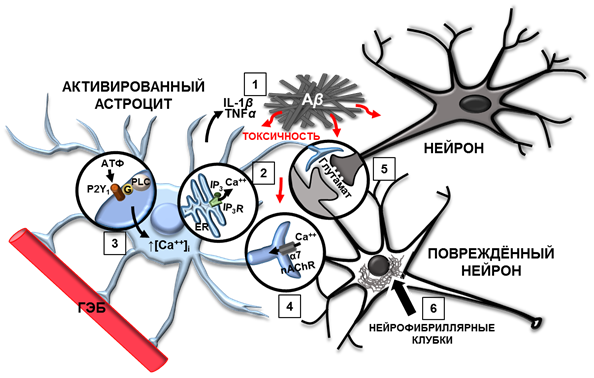
Рис. 1. Изменения в работе астроцитов при болезни Альцгеймера
1. Активированные астроциты продуцируют провоспалительные цитокины: IL-1 и TNFα в ответ на образование Aβ. 2. Передача кальциевых сигналов в астроцитах происходит с помощью вторичного посредника IР3, который связывается с кальциевым каналом на мембранах ЭР и способствует высвобождению ионов Сa2+. 3. Активированные астроциты вблизи амилоидных бляшек экспрессируют метаботропные пуринергические рецепторы P2Y1 до высокого уровня, что приводит к чрезмерному повышению внутриклеточного Сa2+. 4. Aβ индуцирует спонтанное повышение [Сa2+]i в астроцитах гиппокампа посредством α7-никотиновых рецепторов. 5. Пониженная способность к поглощению глутамата у активированных астроцитов влияет на поглощение перисинаптического глутамата, приводя к глутаматной эксайтотоксичности. 6. Внутриклеточные нейрофибриллярные клубки нарушают аксональный транспорт и синаптический метаболизм, приводя к деградации нейрона.
Динамика кальция в астроцитах при болезни Альцгеймера
Ионы кальция – универсальные внутриклеточные посредники, способные регулировать огромное разнообразие внутриклеточных процессов: экзоцитоз, встраивание рецепторов в мембрану, запуск синтеза белков; имеют опосредованное влияние на процессы обучения и памяти [23]. Поступление кальция из внеклеточного пространства происходит в основном через вольтаж-зависимые кальциевые каналы (voltage-gated calcium channel, VGCC), депо-зависимый вход кальция (store-operated Ca2+ entry, SOCE) и Na+/Ca2+ обменники (sodium-calcium exchanger, NCX) [30]. Кальций-связывающие внутриклеточные ферменты имеют различное сродство и чувствительность к ионам Сa2+ (от сотен нМ в цитозоле до концентраций в диапазоне 100–1000 мкМ в ЭР) [23]. Внутриклеточные Сa2+-датчики локализованы в разных частях клетки, поэтому локальные кальциевые градиенты могут независимо друг от друга регулировать отдельные комплексы Сa2+-зависимых процессов [28]. Механизм передачи кальциевых сигналов включает активацию рецепторов, связанных с G-белками, что приводит к вовлечению фосфолипазы C в гидролиз фосфатидил-инозитол-4,5-бисфосфата – фосфолипида клеточных мембран, с образованием вторичных мессенджеров – водорастворимого IР3 и мембранно-связанного диацилглицерола (DAG). IР3 переходит в цитозоль, где связывается со своим рецептором (кальциевым каналом) на мембранах ЭР и способствует высвобождению ионов Сa2+ [31, 32]. Индуцированное G-белками увеличение цитозольного кальция в астроцитах может происходить как в виде колебаний, так и длительным повышением концентрации [30].
Многие исследования подтверждают значительную роль дисрегуляции кальция в развитии БА. Воздействие Aβ индуцирует спорадические Сa2+-всплески в астроцитах [41]. Аберрантные флуктуации астроцитарного кальция начинаются вблизи бляшек Aβ и могут распространяться на большие расстояния по всей коре головного мозга. В человеческих астроцитах, выделенных из мозга пациентов с БА посмертно, был отмечен абнормальный уровень экспрессии генов, связанных с кальциевыми сигналами и гомеостазом [30]. Также было показано, что Aβ42 и его токсический фрагмент Aβ25-35 вызывают спонтанные Сa2+-всплески в астроцитах, растущих в смешанных астроглиально-нейронных культурах: Aβ-индуцированные колебания [Сa2+]i продолжались в течение длительного времени и приводили к гибели нейронов, которая наступала в течение 24 часов после введения Aβ в культуру, однако подавление [Сa2+]i колебаний предотвращало гибель нейронов [42]. Кроме того, применение пикомолярных концентраций Aβ42 индуцировало спонтанное повышение [Сa2+]i в астроцитах гиппокампа посредством α7-никотиновых рецепторов (α7nAChRs) [42]. Эти рецепторы представляют собой лиганд-зависимые ионотропные каналы, высокопроницаемые для Са2+ [43]. Показанное агонистическое воздействие Aβ на функциональные астроцитарные α7nAChR делает эти рецепторы потенциальной терапевтической мишенью при БА.
Собственная кальциевая активность астроцитов
Сейчас уже известно, что астроциты не просто поддерживают, но и активно участвуют в обработке нейронной информации в мозге. Это новое восприятие функционирования ЦНС смещает фокус изучения патологии БА только с нейрональных клеток к взаимодействию астроцитов и нейронов. В литературе появляется все больше доказательств того, что астроцитарный вклад в ранние когнитивные нарушения является важным компонентом БА, но на сегодняшний день остается достаточно малоизученным. Прежде всего астроциты реагируют на колебания внутриклеточного кальция, который контролирует сигнальные свойства астроцитов и их способность к обеспечению нейронов. Aβ может нарушать передачу астроцитарных сигналов кальция, что влияет на высвобождение глиотрансмиттеров, которые жизненно важны для связи нейронов и астроцитов, увеличивая вклад последних в патогенез заболевания. Динамика Са2+ представляет собой основу механизма астроцитарной сигнальной передачи и затрагивает почти все характерные особенности, в том числе генетическую причину БА, а ее нарушения приводят к дисфункции астроцитов и подконтрольных им областей головного мозга посредством аберрантных кальциевых сигналов. Расшифровка кальциевых сигналов и поиск соединений, нормализующих дисрегулированные уровни Ca2+ или специфических блокаторов Ca2+-регулируемых патогенных сигнальных каскадов, теоретически могут вернуть физиологическую роль астроцитам и, следовательно, привести к клиническому улучшению.
Энтрохино-гиппокампальная цепь сильно подвержена болезни Альцгеймера (AD). Здесь мы демонстрируем, что амилоид-β (Aβ) по-разному воздействует на первичные культивируемые астроциты, полученные из энторинальной коры (EC) и гиппокампа от нетрансгенных контролей и трансгенных мышей 3xTg-AD. Воздействие 100 нМ Aβ приводило к увеличению экспрессии метаботропного рецептора глутамата 5 (mGluR5) и его нисходящего рецептора InsP3 типа 1 (InsP3R1) в гиппокампе, но не в EC астроцитах. Амплитуды ответов Ca2 + на агонист mGluR5, DHPG и на АТФ, другой метаботропный агонист, связанный с InsP3Rs, были значительно увеличены в Aβ-обработанном гиппокампе, но не в EC астроцитах. Ранее мы продемонстрировали, что формирование старческого бляшка у мышей 3xTg-AD вызывает астроглиоз в гиппокампе, но не в EC астроцитах. Различные чувствительности инструментария сигнализации Ca2 + от EC против астроцитов гиппокампа до Aβ могут объяснять отсутствие астроглиоза в ЕС, что, в свою очередь, может объяснить более высокую уязвимость этого региона к AD.
Астроциты являются основными гомеостатическими клетками центральной нервной системы. Роль астроглии в патогенезе болезни Альцгейма (AD) остается в целом неизвестной, хотя интерес к астроглиальному ремоделированию в ходе нейродегенерации значительно увеличился в течение последнего десятилетия.1, 6, 7, 8, 9 AD считается одним из наиболее распространенных форм деменции у людей 10, симптомы которых проявляются в прогрессирующих когнитивных нарушениях. Гистопатологические изменения, связанные с АД, включают сенильные бляшки, нейрофибриллярные клубочки, нейровоспламенение, измененную синаптическую связь и гибель нейронов.11, 12 Потеря синапсов, в частности, считается самой ранней ассоциированной с AD клеточной патологией, ответственной за ранние признаки когнитивной недостаточности .13, 14 Эта потеря синапсов может отражать функциональное падение астроцитов, которые, как правило, отвечают за синаптическое поддержание и гомеостаз ионов и нейротрансмиттеров, причем последнее имеет решающее значение для синаптической передачи. Действительно, последние данные указывают на атрофические изменения в астроглии на ранних стадиях AD-подобной патологии у трансгенных модельных животных, что может объяснять недостаточную синаптическую связь.9, 15, 16. На более поздних стадиях, появление β-амилоидных отложений и образование старческих бляшек провоцируют реактивный астроглиоз, который происходит главным образом в клетках, непосредственно связанных с β-амилоидными бляшками.15, 16
Пространственно-временная прогрессия AD хорошо идентифицирована, а энторинальная кору (EC) является самой первой областью, на которую может повлиять патология.17 Нейроны в поверхностных слоях ЕС иннервируют все субрегионы гиппокампа, включая зубчатую извилину (DG) , CA3, CA1 и субикулум через перфорантный путь; синаптические связи между мшистыми волокнами DG и CA3 нейронов и коллатералями Шаффера от CA3 до CA1-нейронов завершают схему прямого гиппокампа.18 В AD значительная потеря нейронов в слое II ЕС происходит на ранних стадиях.19 У пациентов с AD, EC может быть областью, из которой патологический процесс распространяется на другие области мозга; это изначально предполагалось, потому что экспрессия APP, предшественника β-амилоида, была выше в нейронах EC-слоя II, чем в других областях мозга20. Более того, APP, синтезированный EC-нейронами, транспортируется через перфорантный путь до пресинаптических терминалов в DG. В DG наблюдалось глубокое снижение β-амилоидной нагрузки после одностороннего разрыва перфорантного пути.21
Наш предыдущий морфологический анализ22 обнаружил замечательную разницу в AD-ассоциированном поведении астроцитов в ЕС. В отличие от гиппокампа, где реактивные астроциты были характерной особенностью отложений β-амилоидов, в астроцитах EC не проявлялась никакой реактивности и конкретно не ассоциировалась с β-амилоидом. В этом исследовании мы проанализировали сигнализацию Ca2 + и метаботропный молекулярный инструментарий для передачи сигналов Ca2 + в EC и гиппокампальные астроциты in vitro в культурах клеток. Мы обнаружили, что эти астроглиальные популяции имеют отчетливые реакции на лечение β-амилоидом, которые могут объяснять различия в их реактивности в патологическом контексте.
Морфология астроглиальных клеток из EC и гиппокампа, поддерживаемая in vitro в течение первых 3-5 дней, была характерна иная. В культурах гиппокампа большинство клеток были крупными и плоскими (или звездообразными), тогда как в культурах EC многие клетки были тонкие и продолговатые с почти иглообразной морфологией. На рисунке 1а показаны различия в морфологии между культивируемыми астроцитами, полученными из гиппокампа и астроцитов, полученных из ЕС. Оба типа клеток были положительными к иммуноокрашиванию глиальных фибриллярных кислых белков (GFAP), подтверждая тем самым их астроглиальную природу (рис. 1b). Морфология EC астроцитов, поддерживаемая в культуре, была похожа на их появление in situ (рис. 1c) .22 Количественное определение количества тонких и продолговатых клеток показывает значительно более высокий процент этого морфологического профиля в культурах EC (рис. 1a). Общие морфологические особенности астроцитов у не-Tg и 3xTg-AD мышей не выявили существенных различий (рис. 1b).
Но лучше всего, конечно, вообще избежать болезни. Поэтому наряду с разработками лекарств ученые и врачи ищут методы предотвращения заболеваний. Эксперты анализируют образ жизни, состояние здоровья больших групп людей по всему миру и показатели их заболеваемости тем или иным недугом. Недавно в авторитетном международном научно-медицинском журнале Frontiers in neurology вышел обзор данных о профилактике болезни Альцгеймера и деменции (слабоумия). Работу провела крупная группа ученых из Китая , США , Великобритании , Израиля и Канады .
ЧТО ТАКОЕ ПРОФИЛАКТИКА НА САМОМ ДЕЛЕ
Чтобы понять, на каком этапе и кому может помочь соблюдение правил (см. ниже), важно знать, что такое профилактика на самом деле, поясняют авторы работы. Всемирная организация здравоохранения выделяет три вида профилактики:
- первичная профилактика направлена на то, чтобы избежать возникновения болезни;
- вторичная предназначена для того, чтобы распознавать болезнь на ранних стадиях, когда еще даже не появились симптомы. Задача — остановить или замедлить развитие болезни. Этот вид профилактики включает в себя, в том числе, скрининги. То есть проверки и обследования людей из групп риска развития той или иной болезни;
- третичная профилактика применяется, когда человек уже заболел. В данном случае стоит цель избежать прогрессирования болезни, тяжелых осложнений и инвалидности.
Итого: в отличие от бытующих представлений профилактика возможна и тогда, когда человек уже болен. Скажем, ему поставлен диагноз болезнь Альцгеймера. А это значит, что правила, о которых рассказывается ниже, способны помочь абсолютно любому человеку. Если не избежать болезни, то как минимум снизить риск тяжелых осложнений.
ТРИ СТАДИИ АЛЬЦГЕЙМЕРА
Национальный институт исследований старения (США) и Альцгеймеровская ассоциация в 2011 году разработали модель, выделяющую три стадии болезни. На первой стадии у пациента нет ни малейших внешних признаков заболевания, в то время как в мозге уже появляются предшественники грядущей катастрофы. Стартует процесс накопления амилоидных бляшек и деформированного тау -белка, которые впоследствии буквально душат клетки мозга нейроны. Справедливости ради отметим: для исследователей еще остается немало загадок. В частности, амилоидные сгустки-бляшки с помощью метода позитронно-эмиссионной томографии мозга можно найти у многих людей уже с 40 лет. Но, к счастью, болезнь Альцгеймера при этом развивается далеко не у всех. Потому-то ранняя диагностика недуга по-прежнему остается одной из главных проблем.
СПАСИ СЕБЯ САМ
1. Отказаться от курения.
4,7 миллионов случаев заболевания болезнью Альцгеймера во всем мире связаны в первую очередь с курением, посчитали исследователи. Как такое может быть? Если пояснять упрощенно, то вредные токсины, которые попадают в организм при курении, вызывают воспаление стенок кровеносных сосудов. В том числе в головном мозге. Из-за этого ускоренно накапливаются те самые опасные амилоидные бляшки, из-за которых гибнут нервные клетки нейроны.
Если сократить уровень курения в мире на 25%, то удалось бы спасти от Альцгеймера примерно 1 миллион людей, уверяют ученые.

2. Избавиться от лишнего веса и ожирения в среднем возрасте (после 40 лет).
Речь идет о тех, у кого индекс массы тела (ИМТ) выше 30, уточняют ученые. Напомним: чтобы посчитать ИМТ, берем свой рост в метрах, возводим в квадрат. А потом массу тела в кг делим на полученную цифру.
Около 677 000 случаев болезни Альцгеймера в мире связаны главным образом именно с избыточным весом, говорится в исследовании.
В том числе из-за снижения такой защиты начинают ускоренно развиваться процессы, вызывающие болезнь Альцгеймера.
3. Заниматься физическими упражнениями, не вести сидячий образ жизни.
4,3 млн случаев болезни Альцгеймера связаны с недостатком физнагрузок, ужасаются ученые. Из-за этого страдает кровообращение в мозге. А значит — выше риск накопления опасных веществ, которые могли бы вымываться с током крови.
Совет: золотым международным стандартом на сегодня считается минимум 150 минут двигательной активности в неделю, или не менее чем по 30 минут 5 дней в неделю. А лучше — по 40 — 60 минут каждый день. Самый безопасный вид физнагрузок — кардиологическая ходьба, то есть максимально быстрым шагом, но так, чтобы не было одышки.
4. Повышать свое образование.
Низкий уровень образования способствует развитию болезни Альцгеймера у 6,5 млн человек во всем мире, утверждают ученые (как оценивать свой уровень образования — не уточняется).
- Для профилактики болезни Альцгеймера важно создавать так называемый когнитивный резерв. Этого можно добиться с помощью занятий, которые ведут к образованию новых связей между нейронами и стимулируют образование новых нервных клеток (у взрослого человека их появляется очень мало, но тем не менее), - поясняет профессор Яшин . - А самое мощное средство для создания когнитивного резерва — это как раз получение незнакомых навыков и знаний. Например, изучение иностранного языка, посещение увлекательных образовательных лекций или их просмотр по телевизору, овладение компьютерными навыками в пожилом возрасте, обучение танцам, рисованию, лепке — все, что требует от нас запоминания и вспоминания, творческого подхода — все это помогает создавать и укреплять связи между нейронами.
5. Вовремя диагностировать и лечить сахарный диабет.
Примерно 825 000 случаев развития болезни Альцгеймера ассоциируется с сахарным диабетом, пишут авторы исследования.
6. Уделять внимание лечению депрессии.
Это заболевание провоцирует не менее 3,6 млн случаев болезни Альцгеймера во всем мире, предупреждают ученые.
Наличие депрессии само по себе уже сигналит, что в мозге какие-то неполадки, из-за которых нейроны могут оказаться более беззащитны перед повреждающими процессами. Долговременные наблюдения за большими группами людей показывают, что депрессия является важным фактором риска, повышающим вероятность болезни Альцгеймера.
7. Нормализовать давление и не допускать усугубления гипертонии в среднем возрасте (после 40 лет).
По данным авторов исследования из-за нелеченой гипертонии зарабатывают себе Альцгеймер 1,7 млн пациентов. Повышенное давление грозит не только инфарктами и инсультами, но и развитием деменции, подчеркивают ученые.
В ТЕМУ
Энтрохино-гиппокампальная цепь сильно подвержена болезни Альцгеймера (AD). Здесь мы демонстрируем, что амилоид-β (Aβ) по-разному воздействует на первичные культивируемые астроциты, полученные из энторинальной коры (EC) и гиппокампа от нетрансгенных контролей и трансгенных мышей 3xTg-AD. Воздействие 100 нМ Aβ приводило к увеличению экспрессии метаботропного рецептора глутамата 5 (mGluR5) и его нисходящего рецептора InsP3 типа 1 (InsP3R1) в гиппокампе, но не в EC астроцитах. Амплитуды ответов Ca2 + на агонист mGluR5, DHPG и на АТФ, другой метаботропный агонист, связанный с InsP3Rs, были значительно увеличены в Aβ-обработанном гиппокампе, но не в EC астроцитах. Ранее мы продемонстрировали, что формирование старческого бляшка у мышей 3xTg-AD вызывает астроглиоз в гиппокампе, но не в EC астроцитах. Различные чувствительности инструментария сигнализации Ca2 + от EC против астроцитов гиппокампа до Aβ могут объяснять отсутствие астроглиоза в ЕС, что, в свою очередь, может объяснить более высокую уязвимость этого региона к AD.
Астроциты являются основными гомеостатическими клетками центральной нервной системы. Роль астроглии в патогенезе болезни Альцгейма (AD) остается в целом неизвестной, хотя интерес к астроглиальному ремоделированию в ходе нейродегенерации значительно увеличился в течение последнего десятилетия.1, 6, 7, 8, 9 AD считается одним из наиболее распространенных форм деменции у людей 10, симптомы которых проявляются в прогрессирующих когнитивных нарушениях. Гистопатологические изменения, связанные с АД, включают сенильные бляшки, нейрофибриллярные клубочки, нейровоспламенение, измененную синаптическую связь и гибель нейронов.11, 12 Потеря синапсов, в частности, считается самой ранней ассоциированной с AD клеточной патологией, ответственной за ранние признаки когнитивной недостаточности .13, 14 Эта потеря синапсов может отражать функциональное падение астроцитов, которые, как правило, отвечают за синаптическое поддержание и гомеостаз ионов и нейротрансмиттеров, причем последнее имеет решающее значение для синаптической передачи. Действительно, последние данные указывают на атрофические изменения в астроглии на ранних стадиях AD-подобной патологии у трансгенных модельных животных, что может объяснять недостаточную синаптическую связь.9, 15, 16. На более поздних стадиях, появление β-амилоидных отложений и образование старческих бляшек провоцируют реактивный астроглиоз, который происходит главным образом в клетках, непосредственно связанных с β-амилоидными бляшками.15, 16
Пространственно-временная прогрессия AD хорошо идентифицирована, а энторинальная кору (EC) является самой первой областью, на которую может повлиять патология.17 Нейроны в поверхностных слоях ЕС иннервируют все субрегионы гиппокампа, включая зубчатую извилину (DG) , CA3, CA1 и субикулум через перфорантный путь; синаптические связи между мшистыми волокнами DG и CA3 нейронов и коллатералями Шаффера от CA3 до CA1-нейронов завершают схему прямого гиппокампа.18 В AD значительная потеря нейронов в слое II ЕС происходит на ранних стадиях.19 У пациентов с AD, EC может быть областью, из которой патологический процесс распространяется на другие области мозга; это изначально предполагалось, потому что экспрессия APP, предшественника β-амилоида, была выше в нейронах EC-слоя II, чем в других областях мозга20. Более того, APP, синтезированный EC-нейронами, транспортируется через перфорантный путь до пресинаптических терминалов в DG. В DG наблюдалось глубокое снижение β-амилоидной нагрузки после одностороннего разрыва перфорантного пути.21
Наш предыдущий морфологический анализ22 обнаружил замечательную разницу в AD-ассоциированном поведении астроцитов в ЕС. В отличие от гиппокампа, где реактивные астроциты были характерной особенностью отложений β-амилоидов, в астроцитах EC не проявлялась никакой реактивности и конкретно не ассоциировалась с β-амилоидом. В этом исследовании мы проанализировали сигнализацию Ca2 + и метаботропный молекулярный инструментарий для передачи сигналов Ca2 + в EC и гиппокампальные астроциты in vitro в культурах клеток. Мы обнаружили, что эти астроглиальные популяции имеют отчетливые реакции на лечение β-амилоидом, которые могут объяснять различия в их реактивности в патологическом контексте.
Морфология астроглиальных клеток из EC и гиппокампа, поддерживаемая in vitro в течение первых 3-5 дней, была характерна иная. В культурах гиппокампа большинство клеток были крупными и плоскими (или звездообразными), тогда как в культурах EC многие клетки были тонкие и продолговатые с почти иглообразной морфологией. На рисунке 1а показаны различия в морфологии между культивируемыми астроцитами, полученными из гиппокампа и астроцитов, полученных из ЕС. Оба типа клеток были положительными к иммуноокрашиванию глиальных фибриллярных кислых белков (GFAP), подтверждая тем самым их астроглиальную природу (рис. 1b). Морфология EC астроцитов, поддерживаемая в культуре, была похожа на их появление in situ (рис. 1c) .22 Количественное определение количества тонких и продолговатых клеток показывает значительно более высокий процент этого морфологического профиля в культурах EC (рис. 1a). Общие морфологические особенности астроцитов у не-Tg и 3xTg-AD мышей не выявили существенных различий (рис. 1b).
Читайте также:
