
Атрофия зрительных нервов наследуется рецессивно сцеплено с хромосомой
Известно несколько форм наследственных атрофии зрительного нерва, отличающихся друг от друга клиническими проявлениями, характером функциональных нарушений, временем начала заболевания, типом наследования. Лечение наследственных атрофии зрительного нерва должно быть направлено на улучшение трофики; как правило, оно малоэффективно.
Юношеская наследственная атрофия зрительного нерва - двустороннее заболевание с аутосомно-доминантным типом наследования. Встречается чаще других наследственных атрофии и является наиболее доброкачественной формой. Первые офтальмоскопические признаки появляются в 2—3 летнем возрасте, функциональные нарушения наступают значительно позже (в 7-20 лет). Острота зрения снижается постепенно, длительное время остается достаточно сохранной, составляя 0,1—0,9. Появляются центральные и парацентральные скотомы, увеличивается слепое пятно. Концентрическое сужение поля зрения наблюдается редко. Нарушения цветового зрения, как правило, предшествуют снижению остроты зрения. Сначала снижается чувствительность к синему цвету, затем - к красному и зеленому; может развиться полная цветослепота. Темновая адаптация не изменяется. Электроретинограмма, как правило, в норме. Заболевание может сопровождаться нистагмом и неврологическими нарушениями.
Врожденная, или инфантильная, наследственная аутосомно-рецессивная атрофия зрительного нерва встречается реже, чем доминантная форма, проявляется, как правило, при рождении или в раннем возрасте (до 3 лет). Атрофия двусторонняя, полная, стационарная. Острота зрения резко снижена, поле зрения концентрически сужено. Имеется дисхроматопсия. Электроретинограмма в норме. Обычно наблюдается нистагм. Общие и неврологические расстройства встречаются редко. Заболевание следует дифференцировать от гипоплазии диска, инфантильной формы тапеторетинальной дегенерации.
Атрофия зрительного нерва, связанная с полом, встречается редко, проявляется в раннем возрасте и медленно прогрессирует. Острота зрения снижается до 0,4-0,1. Периферические отделы поля зрения сохранены, слепое пятно несколько увеличено. В ранних стадиях заболевания (в молодом возрасте) электроретинограмма в норме, впоследствии снижается и исчезает волна b. Атрофия зрительного нерва может сочетаться с умеренными неврологическими нарушениями.
Осложненная инфантильная наследственная атрофия зрительного нерва Бера чаще передается по рецессивному типу, реже — по доминантному. Начинается рано — на 3-10-м году жизни, когда внезапно снижается зрение, затем процесс медленно прогрессирует.
В ранних стадиях заболевания наблюдается легкая гиперемия диска. Впоследствии развивается частичная (с поражением височной половины диска) или полная атрофия зрительного нерва. Острота зрения может снижаться до 0,05—0,2; полная слепота, как правило, не наступает. Имеется центральная скотома при нормальных границах периферического поля зрения. Часто сочетается с нистагмом (50%) и косоглазием (75%). Характерно наличие неврологических симптомов; поражается преимущественно пирамидная система, что сближает эту форму с наследственными атаксиями.
Атрофия (неврит) зрительного нерва Лебера. Начинается внезапно и протекает по типу острого двустороннего ретробульбарно-го неврита. Интервал между поражением одного и другого глаза иногда может достигать 1—6 мес. Чаще болеют мужчины (до 80-90% случаев). Заболевание может появляться в возрасте 5-65 лет, чаще — в 13-28 лет. В течение нескольких дней, реже 2 -4 нед, зрение снижается до 0,1 - счет пальцев у лица. Иногда снижению зрения предшествуют периоды затуманивания, лишь в единичных случаях наблюдаются фотопсии. Нередко отмечается никталопия, больные лучше видят в сумерки, чем днем. В начальном периоде заболевания может отмечаться головная боль. В поле зрения выявляются центральные скотомы, периферия чаще сохранена, электроретинограмма не изменена. Характерна дисхроматопсия на красный и зеленый цвета.
Глазное дно может быть нормальным, иногда отмечается легкая гиперемия и небольшая нечеткость границ диска зрительного нерва.
Атрофические изменения появляются через 3—4 мес после начала заболевания, сначала в височной части диска. В поздней стадии развивается атрофия зрительного нерва.
У некоторых больных возникают рецидивы или наблюдается медленное прогрессирование процесса, у ряда больных отмечается некоторое улучшение зрительных функций. Неврологические расстройства возникают редко. Иногда отмечаются отклонения на ЭЭГ, нерезко выраженные признаки поражения оболочек и диэнцефальной области.
У членов одной семьи заболевание большей частью протекает однотипно в отношении времени его начала, характера и степени функциональных нарушений. Тип наследования точно не установлен, более вероятна передача по рецессивному типу, сцепленному с полом.
Оптикоотодиабетичеекий синдром - двусторонняя первичная атрофия зрительного нерва, сопровождающаяся резким снижением зрения в сочетании с глухотой неврогенного генеза, гидронефрозом, пороками развития мочевой системы, сахарным или несахарным диабетом. Развивается в возрасте от 2 до 24, чаще до 15 лет.
Наследственные нейрооптикопатии — группа заболеваний, при которых дисфункции зрительного нерва имеют наследственные причины, выявленные на основании семейного наследования или генетического анализа. Клиническая вариабельность одного заболевания как внутри-, так и межсемейная, часто затрудняет распознавание и классификацию этой патологии.
Наследственные нейрооптикопатии часто классифицируются по типу наследования; чаще всего это аутосомно-доминантный, аутосомно-рецессивный и материнский (наследование через митохондриальную ДНК (мтДНК, mtDNA)). Наследуемые по одному типу нейрооптикопатии не обязательно вызываются одним генетическим дефектом. Аналогично, различные генетические дефекты могут являться причиной идентичных или схожих фенотипов — одни из которых передаются по общему типу наследования, другие — нет. И наоборот, один генетический дефект может вызывать развитие различных клинических проявлений, хотя тип наследования должен совпадать.
Также зачастую причиной единичного случая является предполагаемый или подтвержденный генетический дефект, что делает невозможным классификацию на основании типа наследования.
Наследственные нейрооптикопатии обычно манифестируют симметричным двусторонним безболезненным снижением центрального зрения. Многие заболеваний этой группы характеризуются поражением папилломакулярного нервного пучка, что вызывает развитие центральных или центроцекальных скотом. Точная локализация первичного дефекта структур ганглиозных клеток и их аксонов и патофизиологические механизмы поражения зрительного нерва остаются неизвестными, но в большинстве, если не во всех, случаях наследственных нейрооптикопатий центральную роль в патогенезе играет митохондриальная дисфункция. Поражение зрительного нерва обычно персистирует и может прогрессировать. На момент развитии атрофии уже имеется обширное поражение зрительного нерва.
При некоторых наследственных нейрооптикопатиях наблюдается изолированная дисфункция зрительного нерва. При других поражение зрительного нерва всегда сопровождается различными неврологическими и системными аномалиями, некоторые наследственные заболевания, например мультисистемные дегенерации, характеризуются первичными неврологическими или системными нарушениями и могут сопровождаться и атрофией зрительного нерва. В настоящей главе наследственные нейрооптикопатии разделены на три основных группы:
1. Заболевания, преимущественно не сопровождающиеся неврологическими или системными проявлениями.
2. Заболевания, часто сопутствующие неврологическим или системным проявлениям.
3. Состояния, при которых нейрооптикопатия обычно диагностируется как вторичное поражение на фоне общего заболевания.
По мере открытия других специфических генетических дефектов, вероятно, будут изменяться и наши представления о фенотипах и классификации заболеваний этой группы.
Хотя большинство пациентов затрудняются точно определить возраст начала ухудшения зрения, большинство больных отмечают появление жалоб со стороны зрения в возрасте между четырьмя и десятью годами жизни, к одиннадцатилетнему возрасту отмечают ухудшение зрения 58-84% пациентов. Изредка при развитии тяжелого поражения нарушение зрительных функций и сенсорный нистагм наблюдаются уже в дошкольном возрасте. Многие пациенты не подозревают об имеющихся у них нарушениях зрения, пока у них при обследовании членов семьи больного не диагностируется атрофия зрительного нерва.
Такие случаи обычно свидетельствуют о незамеченном дебюте заболевания в детстве, легкой степени зрительной дисфункции, отсутствии ночной слепоты и медленном прогрессировании. Снижение остроты зрения обычно одинаково невелико на обоих глазах. Острота зрения больных варьирует от 20/20 до 20/800, всего лишь около 15% пациентов имеют остроту зрения 20/200 или ниже. Снижение остроты зрения до движения руки или световосприятия встречается крайне редко. Отмечается выраженная межсемейная и внутрисемейная вариабельность тяжести нарушения зрительных функций.
Хотя и не так быстро развивающиеся и не настолько тяжелые, как при наследственной нейрооптикопатии Leber, нарушения зрения при доминантной атрофии зрительного нерва примерно в 50% случаев не позволяют больным управлять механическим транспортным средством. Классическим для больных доминантной атрофией зрительного нерва является нарушение восприятия синего — желтого цветов, но чаще всего наблюдается смешанное нарушение цветовосприятия. При периметрии пациентов с доминантной атрофией зрительного нерва характерно наличие центральных, парацентральных или центроцекальных скотом. Двустороннее снижение чувствительности может симулировать сдавление хиазмы. Атрофия зрительного нерва у пациентов с наследуемой по доминантному типу нейрооптикопатией может протекать незаметно, проявляться лишь побледнением височной половины ДЗН или же вызывать изменения всего диска. Наиболее характерным симптомом является выраженная треугольная экскавация височной половины диска зрительного нерва; иногда таким больным ставится ошибочный диагноз глаукомы.
Незаметное прогрессирующее ухудшение зрительных функций происходит приблизительно у 50-75% пациентов.
Считается, что доминантная атрофия зрительного нерва представляет собой первичную дегенерацию ганглиозных клеток сетчатки. Причиной большинства случаев доминантной атрофии зрительного нерва (50-60%) являются мутации гена ОРА1, локализующегося в длинном плече хромосомы 3 (3q28-q29). Этот ядерный ген широко экспрессируется в митохондриях сетчатки и кодирует динамин-связанный белок, фиксированный на внутренней мембране крист митохондрий. Идентифицировано более двухсот патогенных мутаций гена ОРА1, в том числе миссенс, нонсенс, делеции/инсерции и мутации сплайсинга. Однако моносимптомный фенотип доминантной атрофии зрительного нерва является генетически гетерогенным состоянием, его причиной могут быть дефекты и других локусов. В одной немецкой семье, наследующей доминантную атрофию зрительного нерва, патогенный локус ОРА4 был картирован на хромосоме 18 (18q12.2-12.3), а в двух не родственных семьях из Франции локус ОРА5 был картирован на хромосоме 22 (22q12.1-13.1).
Но ни продукты этих генов, ни их функции описаны не были. Интересно, что при анализе сцепления была выявлена связь глаукомы нормального давления с полиморфизмом гена OPAL.
Классическая картина доминантной атрофии зрительного нерва включает в себя только атрофию зрительного нерва и не сопровождается другими неврологическими или системными нарушениями. Однако давно было известно, что в некоторых семьях у некоторых родственников развивалась классическая картина доминантной атрофии зрительного нерва, а у других больных членов семьи наблюдались другие клинические проявления, такие как сенсоневральная тугоухость и даже хроническая прогрессирующая наружная офтальмоплегия, атаксия, миопатия, периферическая нейропатия и, редко, фенотип, соответствующий наследственной спастической параплегии.
б) Аутосомно-рецессивная атрофия зрительного нерва. Эта форма атрофии зрительного нерва манифестирует при рождении или развивается в раннем возрасте, диагноз обычно ставится в возрасте младше 3-4 лет. Считается, что заболевание наследуется по аутосомно-рецессивному типу, родители пациентов часто являются близкими родственниками. Снижение остроты зрения тяжелое, может развиваться слепота с сенсорным нистагмом. При периметрии определяется сужение полей зрения различной степени, часто выявляются парацентральные скотомы.
Диски зрительных нервов полностью атрофичны, часто формируется тотальная экскавация. По результатам одной офтальмоскопии трудно отдифференцировать это заболевание от инфантильной дегенерации сетчатки, поэтому большое значение для диагностики приобретает электроретинография.
В одной семье французского происхождения, где имелись близкородственные браки, изолированная рецессивная атрофия зрительного нерва была связана с дефектом хромосомы 8 (8q21-q22), патогенный ген был обозначен как ОРА6, хотя продукт гена и его функции еще предстоит определить. В нескольких семьях из Северной Африки с близкородственными браками, наследующих аутосомно-рецессивную атрофию зрительного нерва, изолированную и сопровождающуюся легкой (часто бессимптомной) сенсоневральной тугоухостью, был выявлен дефект гена хромосомы 11, обозначенного ОРА7 (11q14.1—q21), кодирующего трансмембранный белок митохондрий.
в) Х-сцепленная атрофия зрительного нерва. Семейства с подтвержденным сцепленным с полом наследованием атрофии зрительного нерва встречаются крайне редко, особенно редко — наследующие первичную моносимптомную атрофию зрительного нерва. В двух семьях была выявлена сцепленность с одним и тем же локусом Х-хромосомы (Xp11.4-Хр11.2),патогенный ген получил название ОРА2, хотя продукт гена и его функцию еще предстоит установить. В других семьях с предположительно Х-сцепленным типом наследования атрофия зрительного нерва чаще сопровождается другой неврологической и системной патологией.
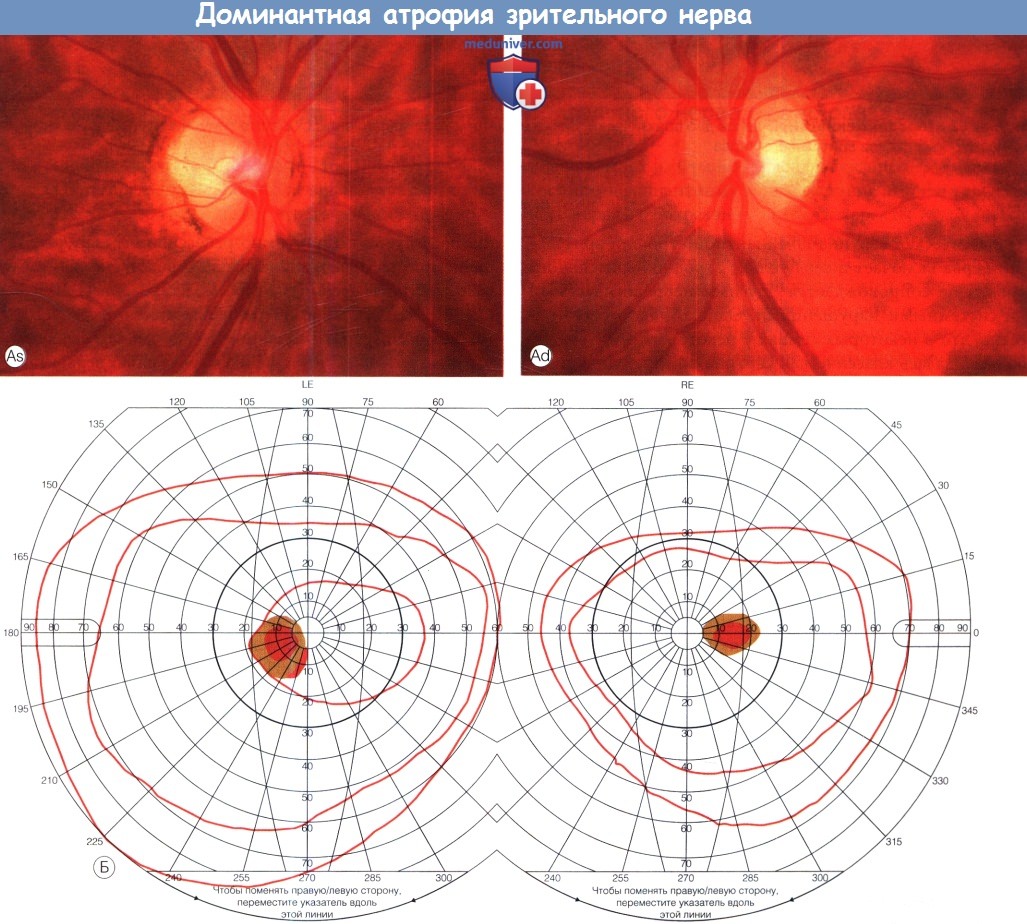
Доминантная атрофия зрительного нерва.
(А) При офтальмоскопии на обоих глазах наблюдаются побледнение височной половины диска зрительного нерва (ДЗН), обширная экскавация.
(Б) При периметрии по Goldmann определяются двусторонние центроцекальные скотомы (снижение чувствительности). 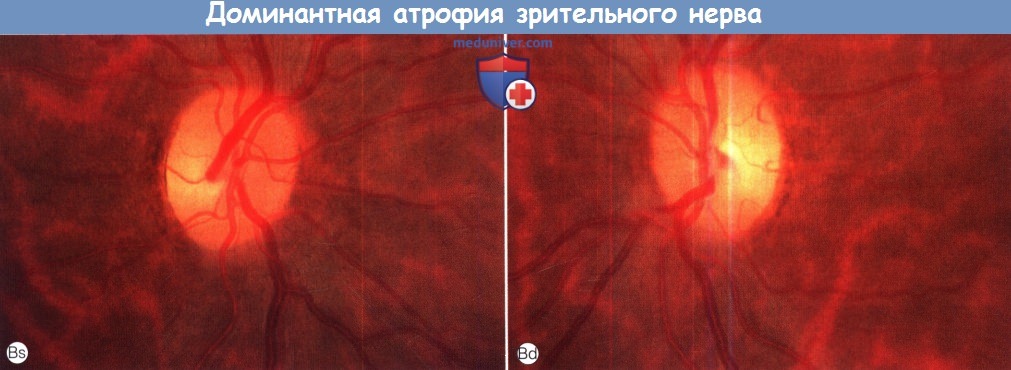
(В) Глазное дно практически здоровой матери того же пациента. Обратите внимание на побледнение височной части обоих дисков зрительного нерва.


Содержание:
- 1 Осложненная форма атрофии зрительного нерва (синдром Бэра)
- 2 Синдром Вольфрама (синдром DIDMOAD)
- 3 Оптикокохлеодентипьный синдром (синдром Ниссена — ван Богарта)
- 4 Инфантильная атрофия зрительного нерва Вента
- 5 Оптикоакустическая атрофия с деменцией Йенсена
- 6 Синдром РАПО(ОАРО)
- 7 Синдром Кен ни — Каффи
- 8 Синдром Розенберга — Чаториана
- 9 Синдром Маринеску — Съегрена
В последнее время аутосомно-рецессивную атрофию зрительного нерва рассматривают как гетерогенную группу заболеваний, общими проявлениями которых являются развивающиеся в раннем возрасте двустороннее снижение зрения и побледнение диска зрительного нерва. H.U.Moller (1991) подверг сомнению существование аутосомно-рецессивной формы простой или врожденной атрофии зрительного нерва, аргументируя свою позицию следующими фактами:
-
с 1968 г. в мировой литературе не описано случаев простой аутосомно-рецессивной атрофии зрительного нерва;
в некоторых сообщениях, появившихся до 1968 г., аутосомно-рецессивное наследование атрофии зрительного нерва предполагалось только на основании того факта, что родители больного состояли в близкородственном браке, а других этиологически значимых причин развития заболевания обнаружить не удавалось;
в ряде случаев при повторном обследовании детей с аутосомно-рецессивной атрофией зрительного нерва диагноз был изменен;
Собственный опыт, основанный на изучении более 1000 случаев атрофии зрительного нерва у детей до 5 лет, обратившихся в консультативно-поликлиническое отделение Тушинской детской клинической больницы и Московского НИИ глазных болезней им. Гельмгольца с 1990 по 2002 г., показывает, что предположения H.U.Moller не лишены оснований. Нам не удалось установить этиологию моносимитом ной атрофии зрительного нерва у 15 % от общего числа обследованных детей. Из этой группы 2,6 % пациентов (0,4 % от общего числа обследованных больных с атрофией зрительного нерва) — дети, родители которых состояли в близкородственном браке. Возможно, что у некоторых больных с врожденной атрофией зрительного нерва неясной этиологии из этой подгруппы заболевание можно было бы рассматривать как вариант аутосомно-рецессивного поражения, но из-за ограниченного числа обследованных членов семей пробандов и отсутствия документированной информации о состоянии глаз у некоторых из родственников в предшествующих поколениях этих родословных не удалось собрать доказательства, необходимые для аргументации существования аутосомно-рецессивного типа наследования.
Таким образом, можно предположить, что простая аутосомно-рецессивная атрофия зрительного нерва — это гетерогенная группа чрезвычайно редко наблюдаемых патологических состояний, для которых характерны двустороннее снижение зрения и деколорация диска зрительного нерва, развивающиеся в первые 4—5 лет жизни.
Необходимо учитывать, что атрофия зрительного нерва может быть одним из клинических проявлений ряда аутосомно-рецессивных системных аномалий, к которым относят:
синдром Вольфрама (Wolfram), или синдром DIDMOAD;
оптикокохлеодентальный синдром, или синдром Ниссена — ван Богар- та (Nissen — van Bogaert);
инфантильная атрофия зрительного нерва Вента (Went);
оптикоакустическая атрофия с деменцией Йенсена (Jensen);
синдром РАПО (GAPO);
синдром Кении — Каффи (Kenny — Caffey);
синдром Розенберга — Чаториана (Rosenberg — Chutorian);
Синдром Бэра относится к гетерогенной группе заболеваний, при которых нисходящая атрофия зрительного нерва сочетается с неврологической симптоматикой (повышенные сухожильные рефлексы, симптом Бабинского, атаксия, спастическая походка, эпилепсия), задержкой психического развития, недержанием мочи. Синдром впервые описан C.Behr в 1909 г. Функциональные нарушения развиваются в течение первых 10 лет жизни. Острота зрении обычно постепенно снижается, но иногда может оставаться стабильной в течение всей жизни. Заболевание часто сопровождается нистагмом и косоглазием.
При морфологических исследованиях обнаружены атрофия зрительных нервов и трактов, выраженная дегенерация ядер латеральных коленчатых тел, изменения в области зрительной лучистости и стриарной коры.
Синдром Вольфрама — чрезвычайно редко наблюдаемое мультисистемное заболевание: частота в популяции 1 : 100 ООО. Заболевание впервые описано D.J. Wolfram и Н.Р. Wage пег (1938). При синдроме Вольфрама атрофия зрительного нерва может сочетаться с несахарным диабетом, глухотой и неврологическими нарушениями (эпилепсия и атаксия).
Снижение остроты зрения обычно начинается в возрасте 5—20 лет. В ранней стадии заболевания определяют дисхроматопсию, а острота зрения нормальная или незначительно снижена. Заболевание характеризуется прогрессирующим течением. Острота зрения снижается от 1,0—0,3 до 0,1—0,02 за период от 3 до 12 лет.
При периметрии выявляют концентрическое сужение периферических границ поля зрения и центральные скотомы.
Синдром Вольфрама наследуется по аутосомно-рецессивному типу. Ген WFS1, ответственный за развитие заболевания, локализуется в 4р16 и кодирует синтез трансмембранного протеина вольфрамина. Нельзя исключить и другие механизмы наследования. Полиморфизм клинических вариантов болезни логичнее объяснить мутациями ядерной или митохондриальной ДНК.
Заболевание впервые описано J. Muller и W. Zeman в 1965 г. Нисходящая атрофия зрительного нерва сочетается с тяжелой неврологической симптоматикой (прогрессирующий спастический тетрапарез), нарастающей тугоухостью и задержкой психического развития. Острота зрения постепенно снижается вплоть до полной слепоты. Заболевание завершается смертью больных в первые 10 лет жизни.
Атрофия зрительного нерва развивается в раннем возрасте, сочетается с пирамидной, экстрапирамидной симптоматикой и ментальным дефицитом. Острота зрения варьирует от 0,4 до 0,002. Регистрируется субнормальная ЭРГ. Возможно, заболевание наследуется по Х-сцеп- ленному типу.
Заболевание проявляется атрофией зрительного нерва, снижением слуха и деменцией. Данная патология развивается в возрасте 5—20 лет и характеризуется прогрессирующим течением.
Акроним РАПО используют для обозначения редко встречающегося аутосомно-рецессивного синдрома, характеризующегося задержкой роста, алопецией, псевдоанодонтией и оптической атрофией (атрофией зрительного нерва). В литературе описано около 10 случаев РАПО. Одним из первых проявлений заболевания является нечеткость границ диска зрительного нерва. Атрофия зрительного нерва характеризуется прогрессирующим течением. При КТ выявляют истончение зрительного нерва в интраорбитальном отделе. Диагноз устанавливают в раннем возрасте вследствие ярко выраженных системных отклонений: отставания в росте, отсутствия зубов и волос на голове, а также краниофациальных аномалий.
Механизмы наследования заболевания изучены недостаточно, впервые описано F.M.Kenny и L.Linarelli (1966), которые сообщили о двух пациентах низкого роста с утолщением трубчатых костей и транзиторной гипокальциемией. Позднее J.Caffey (1967) подробно охарактеризовал рентгенологические признаки заболевания. Офтальмологические изменения наблюдаются у 73 % больных с синдромом Кенни — Каффи и включают гиперметро- ПИК> (50 %), микрофтальм (приблизительно у 70 %), микрокорнеа (40 %), псевдозастой или атрофию зрительного нерва.
В 1967 г. R.N.Rosenberg и A. Chutorial сообщили о двух братьях с заболеванием, характеризовавшимся атрофией зрительного нерва, прогрессирующим полирадикулоневритом, потерей слуха и мышечной атрофией, напоминающей клинически болезнь Шарко — Мари — Тута. Первые проявления заболевания — полирадикулоневрит и нейросенсорная тугоухость, развивающиеся в возрасте 2—6 лет. Снижение зрения и атрофию зрительного нерва диагностируют в возрасте 20 лет. Предполагают, что заболевание наследуется по аутосомно-рецессивному типу или сцеплено с полом.
Синдром Маринеску - Съегрена — редкое мультисистемное заболевание, наследуемое по аутосомно-рецессивному типу. Наиболее часто диагностируемые офтальмологические проявления синдрома — катаракта, атрофия зрительного нерва, нистагм, косоглазие и офтальмоплегия. Основные неврологические симптомы — мозжечковая атаксия, задержка психомоторного развития и миопатия. При КТ и МРТ выявляют атрофию коры мозжечка и ствола мозга.
Электрофизиологические исследования. ЭРГ у пациентов с рецессивной атрофией зрительного нерва обычно нормальная. При регистрации ЗВП в ответ на стимуляцию вспышкой у 90 % пациентов отмечают снижение амплитуды основного позитивного компонента Р100, у 85 % — удлинение еголатентности. При регистрации паттерн-реверсивных ЗВП у всех пациентов определяют снижение амплитуды и удлинение латентности компонента Р100, нарушение конфигурации ответов.
зрелости. Наличие веснушек передается аутосомно-доминантно. Муж болен и гетерозиготен по наличию веснушек, у жены нет веснушек, она здорова, но является носителем гена атрофии зрительных нервов. Могут ли в этой семье родиться: а) здоровые девочки; б) здоровые веснушчатые мальчики; больные мальчики без веснушек

Сцепленное с полом наследование.
b - атрофия зрительных нервов. Сцеплено с Х хромосомой (Х(b)).
А - наличие веснушек.
Муж: АаХ(b)Y
Жена: ааХ(B)Х(b)
P: ааХ(B)Х(b) х АаХ(b)Y
G: аХ(B) aX(b) AX(b) aX(b) AY aY
F1: Аa0Х(B)Х(b) aaX(B)X(b) AaX(B)Y aaX(B)Y AaX(b)X(b) aaX(b)X(b) AaX(b)Y aaX(b)Y
Аa0Х(B)Х(b) - веснушки, девочка здоровая.
aaX(B)X(b) - нет веснушек, девочка здоровая.
AaX(B)Y - Веснушки, мальчик здоровый.
aaX(B)Y - нет веснушек, мальчик здоровый.
AaX(b)X(b) - веснушки, девочка больная.
aaX(b)X(b) - нет веснушек, девочка больная.
AaX(b)Y - веснушки, мальчик больной.
aaX(b)Y - нет веснушек, мальчик больной.
Ответ: в этой семье могут родиться здоровые девочки. Здоровые веснушчатые мальчики могут родиться. Больные мальчики без веснушек могут родиться.
Другие вопросы из категории
1)какие из перечисленных органоидов являются мембранными?
а) лизосомы
б) центриоли
в) рибосомы
г) микротрубочки
д) вакуоли
е) лейкопласты
2) Какие компоненты составляют внутреннюю среду организма человека?
а)секреты желез внутренней и внешней секреции
б)желудочный и кишечный соки
в)спинномозговая жидкость
г)лимфа
д)кровь
е)тканевая жидкость
3)Вегетативная нервная система регулирует деятельность
а)межреберных мышц
б)сердечной мышцы
в)мышц лица
г)мышц конечностей
которых через неделю даст столько же самок. К какому способу относят такое размножение, в чем его особенность? Почему при этом образуются только женские особи? 2)
Читайте также
рова и не является носителем. составьте схему решения задачи определите генотипы родителей возможного потомства пол и вероятность рождения здоровых детей носителей этого гена
ТЕСТ С ВЫБОРОМ НЕСКОЛЬКИХ ПРАВИЛЬНЫХ ОТВЕТОВ
1. Заслугами Г. Менделя является то, что он впервые:
а) разработал основной метод генетики – метод гибридологического анализа;
б) изучил наследование признаков, гены которых находятся в одной хромосоме;
в) установил основные закономерности наследования признаков;
г) доказал зависимость между условиями среды и генотипом организма;
д) изучил наследование признаков, гены которых находятся в разных хромосомах;
е) разработал основные положения хромосомной теории наследственности.
ОТВЕТ - АВД
2. При моногибридном скрещивании исходные родительские формы должны:
а) относиться к разным видам;
б) относиться к одному виду;
в) быть гомозиготными;
г) отличаться по одной паре признаков;
д) быть гетерозиготными;
е) отличаться по нескольким парам признаков.
ОТВЕТ - БВГ (?)
3. Гомогаметным мужской пол является у:
а) двукрылых насекомых;
б) млекопитающих;
в) пресмыкающихся;
г) некоторых чешуекрылых;
д) птиц;
е) ракообразных.
4. По типу комплементарности происходит наследование:
а) окраски цветков у душистого горошка;
б) окраски шерсти у кроликов;
в) групп крови у человека;
г) цвета кожи у человека;
д) окраски оперения у кур;
е) окраски чешуй у лука.
ОТВЕТ: А точно, Б (?)
5. При аутосомно-доминантном типе наследования:
а) признак встречается у мужчин и у женщин;
б) родители обычно здоровы;
в) аномалия проявляется практически в каждом поколении;
г) вероятность рождения ребенка с аномалией – 50%;
д) часто болен один из родителей;
е) вероятность рождения ребенка с аномалией – 25%.
6. В Х-хромосоме человека находятся гены, определяющие развитие таких аномалий, как:
а) гемофилия;
б) альбинизм;
в) карликовость;
г) дальтонизм;
д) атрофия зрительного нерва;
е) гипертрихоз.
ОТВЕТ здесь тоже точно а
сина з дефективним зором та ймовірність народження наступної дитини з патологією.
ожидать дигетерозиготной женщины и мужчины гипертрихозом, дальтонизмом и куриной слепотой.
стекловидное тело сетчатку 6) жёлтое пятно В2. В полости среднего уха находятся косточки молоточек 4) стремечко подковка 5) уздечка наковальня 6) улитка В3. Чувство осязания даёт информацию о таких свойствах предмета, как размер 4)вкус цвет 5)запах форма 6) температура Установите соответствие между содержанием первого и второго столбцов. В4. Установите соответствие между анализаторами и их струй' турами. СТРУКТУРЫ АНАЛИЗАТОРЫ А) стекловидное тело 1) зрительный Б) улитка 2) пространственный (вестибулярный) В) колбочки 3) слуховой Г) палочки Д) наковальня Е) полукружные каналы А Б В Г Д Е В5. Установите соответствие между частями глаза и структура ми, их составляющими. ЧАСТИ ГЛАЗА СТРУКТУРЫ А) веки 1) вспомогательный аппарат глаза Б) зрачок 2) глазное яблоко В) слёзные железы Г) стекловидное тело Д) роговица Е) ресницы А Б В Г Д Е В6.Установите соответствие между анализатором и долей ко ры больших полушарий, в которой осуществляется анализ данных ощущений. АНАЛИЗАТОРЫ ДОЛЯ КОРЫ А) вкусовой 1) височная Б) обонятельный 2) теменная В) зрительный 3) затылочная Г) мышечный Д) кожный (тактильный) А Б В Г Д Установите правильную последовательность биологических процессов, явлений, практических действий. В7. Установите последовательность этапов прохождения света, а затем нервного импульса в глазе и зрительном анализа торе. зрительный нерв Е) хрусталик Б) стекловидное тело Ж) зрительная зона коры В) роговица больших полушарий Г) палочки и колбочки Д) хрусталик В8. Установите последовательность прохождения звука и нервного импульса. А) барабанная перепонка Б) слуховой нерв В) молоточек Г) перепонка овального оконца Д) наковальня Е) наружный слуховой проход Ж) ушная раковина З) улитка И) височная доля КБП К) стремечко
Читайте также:
