
Что такое семейная дизавтономия рейли-дея
Рубрика МКБ-10: G90.1
Содержание
- 1 Определение и общие сведения
- 2 Этиология и патогенез
- 3 Клинические проявления
- 4 Семейная дизавтономия (Райли-Дея): Диагностика
- 5 Дифференциальный диагноз
- 6 Семейная дизавтономия (Райли-Дея): Лечение
- 7 Профилактика
- 8 Прочее
- 9 Источники (ссылки)
- 10 Дополнительная литература (рекомендуемая)
- 11 Действующие вещества
Семейная дизавтономия
Синонимы: синдром Райли-Дея, наследственная сенсорная и автономная невропатия 3 типа, наследственная сенсорно-вегетативная невропатия 3 типа
Семейная дизавтономия - семейная врожденная вегетативная дисфункция, проявляющаяся множественными надсегментарными и сегментарно-периферическими вегетативными нарушениями и расстройствами функций вегетативной нервной системы.
Наследственная сенсорно-вегетативная невропатия 3 типа поражает исключительно для лиц восточноевропейской еврейской популяции, с заболеваемостью 1 на 3600 живорождений. Синдром Райли-Дея затрагивает и мальчиков и девочек. Семейная дизавтономия передается как аутосомно-рецессивный признак.
Описали заболевание в 1959 г. американские врачи C. Riley (род. в 1913 г.) и R. Day (род. в 1905 г.). В классификации J. Dyck (1983) синдром обозначается как наследственная сенсорновегетативная невропатия (НСВН), тип III.
Ген HSAN3 (IKBKAP) локализован на длинном плече хромосомы 9 (9q31).
Уже в раннем детском возрасте возникают трудности при кормлении, временами рвота, склонность к инфекционным заболеваниям верхних дыхательных путей, рецидивирующая пневмония. Характерны эмоциональная лабильность, снижение слезо- и слюноотделения, вестибулопатия, гипергидроз, эритемы, сухожильная гипо- или арефлексия, гипалгезия, расстройства терморегуляции, вкуса, генерализованное понижение болевой и вкусовой чувствительности, эпизоды артериальной гипертензии и ортостатической гипотензии. Иногда периодическая гипертермия, кератит, язвы роговицы, энурез, костные аномалии, в частности сколиоз. 40% пациентов проявляют циклическую картину вегетитивного кризиса, которые могут возникать ежедневно, еженедельно или ежемесячно и следуют за утренним пробуждением. Возможны дизартрия, атаксия, отставание в интеллектуальном развитии.
Отсутствие аксонной вспышки (после внутрикожного введения гистамина) и отсутствие грибовидных вкусовых сосочков языка являются характерными чертами синдрома Райли-Дея.
Диагноз основан на клиническом выявлении как сенсорной, так и вегетативной дисфункции. Основные критерии включают снижение слезоотделение, отсутствие грибовидных вкусовых сосочков языка, сниженные сухожильные рефлексы и аномальный тест на гистамин. ДНК-диагностика должна быть выполнена для подтверждения диагноза.
Дифференциальный диагноз включает в себя другие наследственные сенсорные и вегетативные невропатии.
Лечение должно быть индивидуально, так как клиническое проявление заболевания значительно варьируется. Лечение главным образом направлен на купирование алакремии, желудочно-кишечную и респираторной дисфункцию, а также лабильности артериального давления.

Синдром Райли-Дея у детей носит также название семейной дизавтономии и наследственной сенсорно-вегетативной невропатии III типа (НСВН III). Это тяжелое, генетически обусловленное заболевание проявляется у ребенка в раннем возрасте. Наиболее распространена болезнь среди евреев ашкенази, однако наследственная предрасположенность может передаваться через несколько поколений. Специфическая профилактика отсутствует, лечение поддерживающее, симптоматическое. Проявления невропатии настолько тяжелы, что 25% погибает в течение первого года жизни. В целом дети с синдромом Райли-Дея при постоянной терапии могут прожить максимум до 35 лет. Из-за невозможности полноценной терапии синдром представляет значительную проблему как для специалистов, так и для самих детей и их родных.
Причины развития болезни у ребенка
Заболевание названо по именам педиатров Райли и Дея, описавший синдром в 1949 г. Болезнь распространена в популяции евреев ашкенази, где встречается с частотой 1:10 000 новорожденных. В местах плотного проживания, общинах частота значительно возрастает из-за риска родственных связей и повышенного количества носителей рецессивного гена.
Аутосомно-рецессивный механизм наследственности синдрома Райли-Дея проявляется при рождении детей: ребенок, оба родителя которого являются носителями дефектного гена, с вероятностью в 25% рождается больным. То есть в семье могут родиться 3 носителя гена и один больной малыш. Однако поверхностная статистика не срабатывает в области генетического наследования. Есть примеры, когда в многодетных семьях все дети являлись здоровыми носителями рецессивного гена, и семьи, где из трех рожденных детей все три оказывались больны.
Патогенез заболевания до сих пор уточняется. Есть гипотезы о влиянии нарушения метаболизма, недостаточном продуцировать норадреналина. Данный базовый нейротрансмиттер синаптических синапсов способен вызывать полиморфные проявления нейропатии.
Альтернативная гипотеза базируется на избыточном накоплении ацетилхолина, что блокирует нервные импульсы, не позволяя достигать мышечных волокон. 95,5% патологических случаев вызваны мутацией гена ГКВКАР на участке 9 хромосомы. Остальные 4,5% связаны с еще более редкими дефектами.
Симптомы болезни у детей

Клиническая картина синдрома является сочетанной: как правило, присутствует дисфагия, частая рвота, пониженная поверхностная чувствительность, вегетативная дисфункция, атаксия, недостаточность секреции слезной жидкости и т. д. 40% случаев сопровождаются проявлениями судорожных пароксизмов и задержкой психомоторного развития детей.
Симптомокомплекс в большинстве случаев проявляется в первые часы после появления на свет. Дети отличаются генерализованной мышечной гипотонией, сниженными рефлексами новорожденного. Ребенок с синдромом Райли-Дея слабо кричит, тихо плачет, слезотечение, появляющееся у детей старше трех месяцев, у ребенка с данным заболеванием отсутствует.
В первое время основными симптомами, которые отмечают родители, становятся возникающие проблемы с питанием: дети с трудом сосут, глотают (дисфагия), отмечается частое, циклическое срыгивание, рвота. При рвоте может начинаться кровотечение слизистой желудка.
Значительные трудности с кормлением сохраняются в течение всего детского возраста. При недостаточности питания ребенок отстает в физическом развитии, начинается гипотрофия. В возрасте от рождения до 5 лет отмечаются эпизоды апноэ, обмороки. В 40% случаев у детей диагностируют эпилептические приступы (генерализованные, тонико-клонические), которые могут сопровождаться повышением температуры тела, кислородной, дыхательной недостаточностью.
Дисфункции вегетатики проявляются в гипергидрозе, обильном слюнотечении, расстройствах терморегуляции с беспричинной гипертермией, повышением артериального давления, цианозом, ощущением холода, трофическими язвами конечностей, диспепсией кишечника и т. д.
Нарушенная чувствительность проявляется снижением или отсутствием реакции на боль и различие в температуре. Ребенок с нарушением поверхностной чувствительности может не чувствовать прикосновение к горячей плите, не замечать порезы, что приводит к бытовому травматизму. С развитием заболевания отмечается присоединение атаксии, влияющей на крупную моторику.
Болезнь во многих случаях влияет на темпы психического развития, умеренно снижая когнитивные навыки. Дети (в особенности девочки) отличаются запаздыванием полового созревания. В дальнейшем отмечают присоединение полиневропатий, прогрессирующего сколиоза.
Дети с синдромом Райли-Дея легко инфицируются, часто болеют, в особенности воспалительными поражениями органов дыхания: трахеобронхитами, воспалениями легких.
Диагностика, лечение, профилактика

Диагностика основывается на симптомокомплексе, проведении гистаминовой пробы и анализа ДНК ребенка. Гистаминовая проба — введение гистамина в растворе внутрикожно, что в норме провоцирует гиперемию, покраснение кожных покровов в месте инъекции диаметром от 1 до 3 см. При синдроме Райли-Дея кожная реакция отсутствует. При необходимости дифференциации диагноза иные патологии (другие врожденные полиневропатии, иммунодефициты) исключаются нейросонографическим или магнитно-резонансным исследованием, на котором при данной болезни не выявляется патологий и отклонений.
Лечения, приводящего к выздоровлению, не разработано, так как нет достоверной информации о патогенезе синдрома. Сегодня комплексная симптоматическая терапия направлена на поддержку организма ребенка и профилактику осложнений заболевания.
Профилактика возникновения болезни также неспецифична. При браке между представителями популяции евреев ашкенази и их потомков рекомендовано прохождение ДНК-теста и консультации у генетика с целью определения вероятного риска развития семейной дизавтономии у плода. Во время беременности проводятся тесты на 12-13 неделе гестации, при необходимости с повторением на 16-18. Иных мер профилактики не разработано. Вероятно, в будущем альтернативная профилактика синдрома, также, как и терапия у детей будет разработана в области генной инженерии.
Поделись статьёй и все узнают, что ты разбираешься в здоровье детей! Спасибо ツ
Семейная дизавтономия, заболевание, известное также под именем синдрома Райли-Дея, — болезнь, которую мы так и не умеем лечить. Чаще всего она встречается у евреев ашкенази, однако из-за особенностей генетического наследования может проявляться и в других популяциях. При семейной дизавтономии дети рождаются уже с нарушениями, однако внешне это незаметно. Как определить симптомы болезни, кто находится в группе повышенного риска и какие перспективы сегодня предлагают ученые, рассказывает MedAboutMe.

Сегодня каждый 30-й в группе ашкенази — носитель рецессивного гена, причины синдрома Рая-Дейли. Ребенок с болезнью рождается, только если этот ген есть у обоих родителей, и в среднем шансы на болезнь у детей в таких парах — 25%. Это, однако, совсем не означает гарантии, что в семье из 4 детей родится только один больной малыш: даже в многодетных семьях часть детей может быть совершенно здорова, часть — стать носителем рецессивного гена, и никто не заболеет. А может быть и наоборот: в семье с тремя детишками все три окажутся больны. Предсказать частоту невозможно.
В среднем среди ашкенази дети с данным синдромом рождаются с частотой 1:10 000 младенцев, у других народов частота намного ниже, но болезнь также встречается. Сегодня благодаря генному тестированию обоих родителей до зачатия частота заболевания постепенно снижается.
Разрушенные протеиновые шоссе

Сложности с кормлением — основной симптом, дисфагия, затрудненное глотание будет сопровождать больного всю жизнь, что приводит к недостаточному набору веса, отставанию в развитии тела, в некоторых случаях — и психики.
Дети требуют постоянных процедур по уходу, поддержке, специального питания. При этом на сегодня прогноз лечения неблагоприятный, максимальный срок жизни — 35 лет.
Варианты и перспективы терапии

Ребенок с семейной дизавтономией должен постоянно наблюдаться у невролога, гастроэнтеролога, офтальмолога и курироваться педиатром. В качестве симптоматической терапии предлагаются противорвотные, гипотензивные препараты, лечебная физкультура, массаж, а также занятия с психологом для обучения управления реакциями нервной системы.
Наиболее активно заболевание исследуют и помогают маленьким пациентам в Израиле: в этой небольшой стране и повышены риски болезни, и количество болеющих детей значительно — более 120 человек. Есть и всеизраильский центр лечения семейной дизавтономии на базе отделения пульмонологии в детской больнице, где проблемой занимаются 25 специалистов различного профиля.

Новое исследование специалистов Тель-Авивского университета подтвердило, что вещество фосфатидилсерин, известное как добавка к различным диетам, помогает больным синдромом Рая-Дейли.
Данное вещество — из группы фосфолипидов, которое мы получаем с пищей (в основном — с мясом, рыбой, белой фасолью). В качестве диетической добавки или БАДа у фосфатидилсерина уже есть довольно богатая история: его результативность подтверждена 16-ю научными исследованиями, показавшими эффект от приема при деменции и нарушении когнитивных функций.
Основная проблема с исследованиями фосфатидилсерина в последние годы заключалась в том, что сначала препарат получали из мозга крупного рогатого скота. А после эпидемии коровьего бешенства перешли на изучение той же химической формулы, получаемой из сои (соевый лецитин).
Новые исследования обнадеживают: и с неизлечимыми болезнями можно жить долго, не теряя в качестве жизни. И тем не менее, вне зависимости от поисков терапии, специалисты советуют всем будущим родителям из группы риска проходить генетические тесты до зачатия ребенка, и всем будущим мамам не пренебрегать скринингами и заботиться о здоровье заранее.
Синдром Райли-Дея — тяжелая, генетически обусловленная сенсорно-вегетативная невропатия. Симптомокомплекс включает сочетание дисфагии, рвоты, пониженной поверхностной чувствительности, вегетативной дисфункции, атаксии, недостаточной секреции слезной жидкости. Диагностировать синдром Райли-Дея помогает проведение гистаминовой пробы и ДНК-анализа, исключение другой патологии при помощи нейросонографии или МРТ. Терапия симптоматическая, направлена на купирование рвоты, возмещение потерь жидкости, нормализацию артериального давления, уменьшение мышечной гипотонии, нейропсихологическую коррекцию.
- Причины синдрома Райли-Дея
- Симптомы синдрома Райли-Дея
- Диагностика синдрома Райли-Дея
- Лечение и прогноз синдрома Райли-Дея
- Цены на лечение
Общие сведения
Причины синдрома Райли-Дея
В настоящее время патогенез семейной дизавтономии не известен. Предполагается, что субстратом заболевания выступают нарушения метаболизма, в частности норадреналина — базового нейротрансмиттера симпатических синапсов. Согласно другой гипотезе, синдром Рейли-Дея связан с избыточным накоплением в нервно-мышечных синапсах ацетилхолина и блокировкой прохождения нервного импульса к мышечным волокнам. Патоморфологически наблюдаются дегенеративные изменения и зоны демиелинизации в синаптических ганглиях, задних столбах и корешках спинного мозга, вестибулярном и тройничном нервах, в ретикулярной формации, стволе головного мозга.
Последние исследования в области генетики позволили установить, что наследственный характер патологии детерминирован генетическим дефектом в локусе 9q31 9-й хромосомы. У 95,5% пациентов выявлен однотипный дефект гена ГКВКАР, у остальных определены еще 2 более редко встречающиеся мутации. Наследуется синдром Рейли-Дея по аутосомно-рецессивному механизму. Если оба родителя являются носителями патологического гена, то вероятность рождения больного ребенка составляет 25%.
Симптомы синдрома Райли-Дея
Как правило, клинические проявления семейной дизавтономии появляются сразу после рождения ребенка. Дети слабо кричат, у них наблюдается генерализованная мышечная гипотония и снижение рефлексов новорожденных. Вначале на первый план выходят проблемы с кормлением: отмечаются затрудненное глотание (дисфагия) и сосание, циклические срыгивания, частые рвоты. На фоне дисфагии может возникнуть аспирационная пневмония. Рвота может спровоцировать желудочное кровотечение; в таких случаях в рвотных массах наблюдается примесь крови. Проблемы с питанием сохраняются на протяжении всего детского периода, могут привести к развитию гипотрофии и отставанию ребенка в физическом развитии. В возрасте до 5 лет у детей наблюдается пароксизмы апноэ, зачастую приводящие к развитию синкопальных состояний (обмороков). У 40% детей отмечаются тонико-клонические генерализованные эпиприступы, которые в ряде случаев ассоциируются с гипертермией или гипоксией, обусловленной дыхательными расстройствами.
Вегетативная дисфункция проявляется гипергидрозом, обильной саливацией, расстройством терморегуляции с периодическими беспричинными подъемами температуры тела (иногда - гипотермией), лабильностью артериального давления (транзиторной артериальной гипертензией, артериальной гипотонией при переходе в вертикальное положение), постоянным похолоданием, цианозом и трофическими изменениями кожи дистальных отделов конечностей, чередованием диареи и запоров, пониженной секрецией слезы. Последний симптом становится очевиден после 3-месячного возраста. В норме в этот период плач начинает сопровождаться слезотечением, а у детей, имеющих синдром Райли-Дея, слезотечение отсутствует или резко снижено. Снижение продукции слезы зачастую приводит к возникновению язвенного кератита. Вследствие расстройства вегетативной иннервации периферических сосудов на коже периодически появляются эритематозные пятна. В возрасте 4-5 лет возникают затяжные вегетативные кризы длительностью до нескольких дней. Они сопровождаются профузным потоотделением, рвотой каждые 25-30 мин., беспокойством, подъемом АД, появлением пятнистой эритемы.
Нарушения чувствительности проявляются отсутствием болевого и температурного восприятия кожи и способности различать несколько одинаковых раздражителей, одновременно воздействующих на разные участки поверхности кожи. Глубокие виды чувствительности (вибрационная, мышечно-суставное чувство) менее нарушены. Сенсорные нарушения способствуют частому травматизму ребенка. С возрастом к вегетативным и сенсорным расстройствам присоединяется атаксия, которая становится заметной, когда ребенок начинает ходить. Походка отличается медлительностью и неуклюжестью, что, вероятно, связано с сенсорными нарушениями.
Зачастую синдром Райли-Дея сопровождается некоторой задержкой психического развития, умеренным снижением когнитивной сферы. У детей (особенно у девочек) наблюдается более позднее половое созревание. Типичны нечеткость и гнусавый оттенок речи. С возрастом к клинической картине заболевания присоединяются полиневропатии. У большинства пациентов развивается прогрессирующий сколиоз. Дети относятся к часто болеющим, поскольку подвержены различным инфекционным процессам. Особенно часто у них возникают воспалительные поражения дыхательных путей: трахеобронхиты и пневмонии.
Диагностика синдрома Райли-Дея
Диагноз базируется на характерном сочетании клинических симптомов (вегетативная дисфункция, сенсорные расстройства, пониженная слезная секреция, рвота, атаксия) и их возникновении в раннем детском возрасте. Для оценки психического развития ребенка назначается нейропсихологическое обследование. Из дополнительных исследований диагностическую ценность представляет гистаминовый тест. В норме внутрикожное введение раствора гистамина вызывает покраснение кожи на 1-3 см вокруг места инъекции. Отсутствие какой-либо реакции подтверждает синдром Райли-Дея.
Аппаратные исследования проводятся с целью исключения другой патологии нервной системы. Грудным детям назначают нейросонографию через родничок, детям старшего возраста — МРТ головного мозга. При семейной дизавтономии данные обследования не выявляют никаких патологических отклонений. Дифференциальный диагноз проводится с другими наследственными полиневропатиями и врожденными иммунодефицитами. Пациентам рекомендована консультация генетика и ДНК-диагностика. Проведение генеалогического исследования подтверждает аутосомно-рецессивный тип наследования.
Лечение и прогноз синдрома Райли-Дея
Специфическая терапия не разработана, поскольку отсутствуют ясные представления о патогенезе заболевания. Возможно, будущее лечение семейной дизавтономии находится в сфере генной терапии. На сегодняшний день осуществляется симптоматическое поддерживающее лечение, которое требует участия сразу нескольких специалистов: невролога, гастроэнтеролога, ортопеда, офтальмолога, педиатра. При рвоте компенсация потерянной жидкости проводится путем инфузионной терапии, в качестве противорвотного средства назначается хлорпромазин. При артериальной гипертензии осуществляется гипотензивная терапия. Для уменьшения мышечной гипотонии и предупреждения развития сколиоза рекомендовано ЛФК, занятия на специальных тренажерах, массаж. При задержке психического развития показана нейропсихологическая коррекция.
К сожалению, синдром Райли-Дея имеет неутешительный прогноз. Около четверти заболевших погибают в раннем детском возрасте в связи с тяжелыми нарушениями питания или интеркуррентными инфекциями преимущественно дыхательных путей. В ряде случаев при комплексной симптоматической терапии и адекватном уходе пациенты доживают до 35-летнего возраста.
Семейная вегетативная дисфункция (синдром Райли-Дея) — аутосомно-рецессивное заболевание, распространенное среди восточно-европейских евреев, в этой популяции заболеваемость составляет 1:10 000-1:20 000, частота носительства аномального гена расценивается как 1 %. Заболевание редко встречается в других этнических группах. Аномальный ген картирован в локусе 9q31-q33.
Семейная вегетативная дисфункция (синдром Райли-Дея) с поражением периферической нервной системы характеризуется уменьшением количества мелких немиелинизированных нервных волокон, переносящих импульсы болевой, температурной и вкусовой чувствительности и относящихся к вегетативной нервной системе. Выявляется также дефицит крупных миелинизированных афферентных нервных волокон, которые проводят импульсы от мышечных веретен и сухожильного комплекса Гольджи. Степень анатомических изменений в периферических и особенно вегетативных нервных волокнах широко варьирует. Грибовидные сосочки языка (вкусовые почки) отсутствуют или их количество уменьшено.
Семейная вегетативная дисфункция (синдром Райли-Дея) дебютирует в младенческом возрасте, первыми симптомами служат слабое сосание и глотание. Возможна аспирационная пневмония. Затруднения при приеме пищи остаются основными клиническими проявлениями заболевания на протяжении всего детства. Возможны приступы рвоты. Частые симптомы — избыточная потливость и пятнистая эритема, особенно во время приема пищи или при возбуждении ребенка. Аффективно-респираторные приступы с последующим развитием синкопальных состояний характерны в первые 5 лет жизни. По мере взросления возникает нечувствительность к боли, приводящая к частым травмам. Характерны изъязвления роговицы.
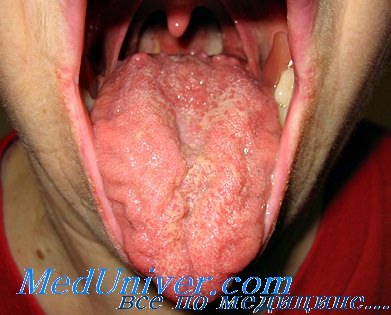
Прорезывание новых зубов приводит к появлению язв на языке. Походка замедленная или неуклюжая, может напоминать атактическую походку в связи с нарушением сенсорного механизма обратной связи посредством мышечных веретен. Атаксия, вероятно, связана в большей степени с нарушением механизма обратной связи посредством мышечных веретен и с дисфункцией вестибулярных нервов, чем с нарушением функции мозжечка. Сухожильные рефлексы отсутствуют.
Сколиоз — тяжелое осложнение, развивающееся у большинства пациентов, обычно прогрессирующего характера. Нормальное слезотечение во время плача у здоровых детей отсутствует до 2-3 мес; у детей с семейной вегетативной дисфункцией слезовыделительная функция не формируется и после этого возраста или значительно нарушена (снижено количество выделяемой слезной жидкости).
Примерно у 40 % пациентов возникают генерализованные тонико-клонические судороги, которые в некоторых случаях ассоциируются с острой гипоксией во время задержки дыхания при аффективно-респираторных приступах, иногда провоцируются высокой лихорадкой, но в большинстве случаев возникают без видимых причин. Контроль температуры тела нарушен; возможна как гипотермия, так и высокая лихорадка. Интеллект обычно снижен, что, однако, не обусловлено эпилепсией. Нередко наблюдается задержка полового созревания (позднее наступление пубертата), особенно у девочек. Речь часто смазанная, нечеткая или имеется носовой оттенок голоса.
После 3-летнего возраста появляются вегетативные кризы, обычно в сочетании с приступами циклической рвоты, продолжающимися 24-72 ч или даже несколько дней. Позывы на рвоту и рвота возникают каждые 25-20 мин в сочетании с гипертензией и профузным потоотделением, появлением пятнистых высыпаний на коже, беспокойством и раздражительностью. Возможно выраженное вздутие желудка, которое вызывает боль в животе и даже дыхательные нарушения. Тяжелые приступы неукротимой рвоты могут осложняться кровавой рвотой (гематемезис).
Синдром Оллгрова — клинический вариант заболевания, для которого характерна алакримия (отсутствие слезоотделения), ахалазия, вегетативная дисфункция в сочетании с ортостатической гипотензией и вариабельным изменением ЧСС, сенсомоторной полиневропатией, обычно с дебютом в подростковом возрасте. Возможна холинергическая дисфункция.
Дизавтономия семейная (Райли - Дея семейная вегетативная дисфункция, Райли - Дея синдром) - наследственное заболевание, проявляющееся диффузным симптомокомплексом вегетативной дисфункции. Встречается преимущественно в еврейской популяции с частотой 1:50. Тип наследования аутосомно-рецессивный. Этиология не выяснена. Полагают, что в основе заболевания лежат нарушения обмена веществ, прежде всего биосинтеза катехоламинов, гомованилиновой и ванилин-миндальной кислот.
Семейная дизавтономия райли-дея. Причины и симптомы
В клинической картине превалируют перманентные и пароксизмальные вегетативные расстройства в сочетании с нарушениями координации. Облигатные симптомы: усиленное потоотделение, нарушение терморегуляции с циклическими подъемами температуры тела, снижение слезоотделения, циклические рвоты, дисфагия, дизартрия, симметричные вегетативно-трофические изменения кожи туловища и дистальных отделов конечностей в виде акроцианоза, пустулезных высыпаний, преходящей пятнистой эритемы, похолодания рук и ног; наблюдаются также снижение кожной болевой и дискриминационной чувствительности, снижение вкуса, отсутствие грибовидных сосочков на языке, расстройства координации. Менее часто встречаются снижение чувствительности роговицы, ее изъязвление, периферические сосудистые расстройства, преходящая артериальная гипертензия, снижение или отсутствие сухожильных рефлексов, приступы апноэ, снижение восприятия запахов, изменения психики, умственная отсталость.
Вопросы и ответы:
Просто напишите в поисковую строку название заболевания (или его часть), либо интересующий лечебный профиль, виде лечения или необходимую процедуру – и система сама предложит наиболее эффективные санатории, пансионаты, спа- и велнес- отели. Вам остается лишь выбрать объект по своим предпочтениям: наиболее оптимальный по цене, расположению, уровню, отзывам или другим параметрам. Все предлагаемые здравницы проходят тщательный предварительный отбор и проверку нашими специалистами и врачами.
Кроме того, можно просто заказать звонок нашего профильного врача-курортолога, который внимательно выслушает Вас, даст свои рекомендации по выбору объекта с наиболее подходящими и эффективными лечебно-оздоровительными программами.
Общий список процедур, предоставляемых в выбранном Вами объекте можно посмотреть на его официальной странице в Здравпродукте.
А вот точный перечень для Вашего лечения скажет только врач в санатории на основе изучения предоставленной санаторно-курортной книжки, первичного осмотра и, возможно, пройденной диагностики и сданных анализов.
Он учтет все Ваши показания и противопоказания, необходимый лечебный профиль, другие пожелания, и назначит максимально эффективный курс лечения исходя из длительности купленной Вами путевки.
Это зависит от вашего основного и сопутствующих диагнозов, выбранной программы и назначений врача санатория с учетом вашего состояния здоровья и рекомендаций лечащего врача, у которого вы проходили обследование.
Для приобретения путевки потребуются:
- Паспорт (свидетельство о рождении - для детей)
- Санаторно-курортная карта (в случае, когда это является обязательным условием)
- Необходимые документы для предоставления скидки, в случае наличия акционных предложений (например, пенсионное удостоверение).
В базе данных ЗдравПродукт® можно найти самые различные санатории и спа-отели по всему миру – какие-то из них являются узкопрофильными (значит специализируются на одном основном лечебном профиле), какие-то – многопрофильными (значит лечат сразу несколько видов заболеваний).
Но здесь важно понимать, что узкопрофильные здравницы – максимально эффективные.
Поэтому наш основной совет – выбирайте, что для вас является самым важным в лечении, а что дополнительным. Исходя из этого, подбирайте объект с вашим основным профилем, и с остальными лечебными профилями в качестве дополнительных.
Кроме того, можно просто заказать звонок нашего профильного врача-курортолога, который внимательно выслушает Вас, даст свои рекомендации по выбору объекта с наиболее подходящими и эффективными лечебно-оздоровительными программами.
Помимо этого, задать свои вопросы и оставить заявку на подбор санатория можно по электронной почте: doctor@zdravproduct.com.
Кроме того, бывают объекты:
- Узкопрофильные и многопрофильные
- Загородные или городские
- Крупные курортные комплексы или небольшие камерные здравницы
- Сетевые или частные
- С собственными источниками и грязями или с привозными
- С собственным медцентром, бальнеоцентром, СПА-комплексом спорткомплексом, или расположенные рядом с большими городскими комплексами или другими объектами
- С открытым бассейном или закрытым, а может и вообще без него
- С пляжем или без
И это многие, но далеко не все возможные отличия.
Читайте также:
