
Дегенеративная болезнь базальных ганглиев неуточненная
Болезнь Паркинсона представляет собой нейродегенеративное заболевание. Касается пожилых людей – около 2/3 больных старше 65 лет, хотя бывают также случаи заболевания до 50 лет. Эта болезнь, медленно и коварно прогрессирующая, неизбежно приводит к постепенному уменьшению физических возможностей, а также интеллектуальных способностей.
Из-за общего старения населения мира, болезнь Паркинсона из года в год становится все более серьезной проблемой, тем более, что современная медицина не знает эффективного лекарства.
Причины болезни Паркинсона
В основе этого заболевания лежит дегенеративный процесс ограниченной группы нейронов (нервных клеток), расположенных среднем мозге, так называемые базальные ганглии, которые играют важную роль в контроле двигательной функции.
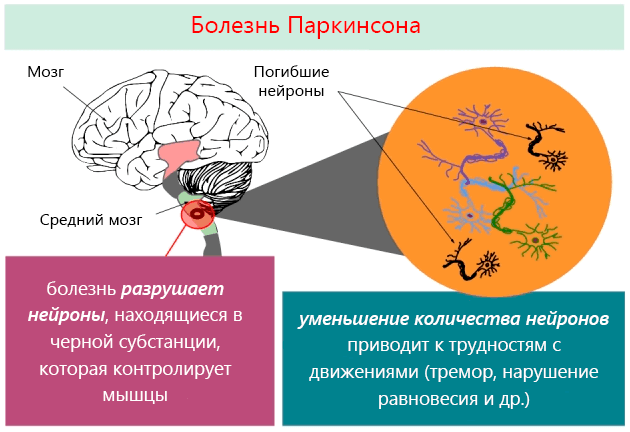
Один из этих ядер, называемое черной субстанцией, вырабатывает и выделяет очень важное вещество – допамин, который играет ключевую роль в контроле процессов движения. При болезни Паркинсона количество клеток черной субстанции снижается, что проявляется прогрессирующим снижением концентрации допамина в базальных ганглиях, что, в свою очередь, ведет к развитию болезни.
Из-за очень больших компенсаторных способностей мозга, симптомы болезни Паркинсона появляются только тогда, когда умрет около 80% клеток. Несмотря на то, что болезнь Паркинсона известна уже много лет, до сих пор не известно, что является причиной, ведущей к дегенерации клеток черной субстанции.
Иногда паркинсонизм может быть связан с использованием препаратов из группы нейролептиков. Это называется медикаментозный паркинсонизм.
Симптомы болезни Паркинсона
Центральная нервная система пациентов с паркинсонизмом нестабильна и её состояние ухудшается с течением времени. Болезнь Паркинсона обычно поражает людей в возрасте старше 60 лет.
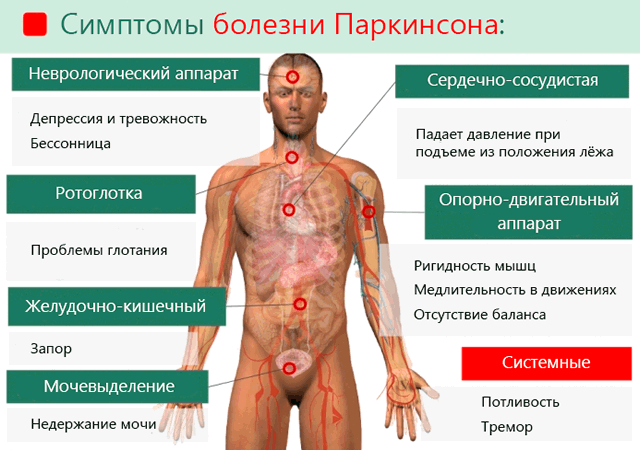
У каждого больного паркинсонизм проявляется немного по-разному. Темпы развития заболевания также индивидуальны. Симптомы болезни делятся на две группы: основные и вторичные.
Жесткие мышцы – это еще один симптом болезни Паркинсона. Часто возникает боль в мышцах, особенно при движении. Жесткое лицо из-за исчезновения мимики, невнятная речь, мелкий и неразборчивый почерк, трудности при глотании.
Болезнь Паркинсона, в целом, затрудняет перемещение, в том числе ходьбу, поэтому трудно начать идти. Часто человек, страдающий от паркинсонизма, останавливается во время движения из-за отказа мышц выполнять команды.
К вторичным симптомам паркинсонизма можно отнести частые запоры связанные с отсутствием контроля над кишечником и мочевым пузырем. Больные испытывают также проблемы с глотанием пищи и слюны.
Нарушается восприятие мира, что приводит к развитию тревоги и депрессии. Ограничивается также моторика, что проявляется шептанием при разговоре, сложностями с письмом и замедленной реакцией на заданный вопрос.
Болезнь Паркинсона вызывает чрезмерное слюноотделение и потоотделение, а также сухость кожи на лице и голове.
Болезнь развивается медленно, вызывая все больше и больше отклонений. Больные умирают, как правило, из-за осложнений, вызванных неподвижностью, таких, как воспаление легких или легочной артерии.
Лечение болезни Паркинсона
Не существует действенного лечения этой болезни. Однако, современная медицина предлагает лекарственные средства, которые позволяют отсрочить на несколько лет появление ярко выраженных симптомов заболевания, увеличивают продолжительность жизни больных почти на время жизни населения в целом и значительно улучшает качество жизни пациента.
К ним относятся:
- Леводопа – препарат, являющийся предшественника допамина;
- Агонисты дофамина (например, бромокриптин, прамипексол) – лекарства, которые имитируют действие допамина;
- Селегилин – препарат, блокирующий моноаминоксидазу типа-B – фермент, который разрушает дофамин.
Для симптоматического лечения важны такие элементы поведения, как:
- диета: должна быть подобрана индивидуально, чтобы не допустить изменения массы тела, содержать правильные пропорции жидкости и клетчатки; кроме того, пациенты, принимающие леводопа должны принимать меньшее количество белка;
- правильный режим жизни;
- упражнения, препятствующие развитию дегенеративных изменений и болевых синдромов;
- интенсивное лечение сопутствующих расстройств, таких, как запор или депрессия.
Пациентов, которые не реагируют на стандартные лекарства, особенно в случае сильного тремора, можно попытаться вылечить, используя один из новых хирургических методов:
- таламотомия – процедура, при которой хирург уничтожает небольшую область структуры мозга, называемой таламус, что приводит к снижению дрожи у 80-90% больных;
- пересадка стволовых клеток – экспериментальная техника, вызывающая много споров, хотя некоторое количество пациентов демонстрирует значительное улучшение, а у некоторых из улучшения настолько велики, что они могут играть в теннис, кататься на лыжах и управлять автомобилем.
Церебральный паркинсонизм также лечат с помощью лекарственных препаратов из группы холинолитиков, которые уменьшают количество ацетилхолина, а точнее, выравнивают зависимость уровня адреналин – ацетилхолина.

Двигательные расстройства возникают в результате дисфункции базальных ганглиев - таламокортикальных моторных сетей нейронов и включают в себя большой спектр двигательных нарушений, от гипокинетических расстройств , например, болезни Паркинсона ( Parkinson) , до гиперкинетических расстройств - болезнь Гентингтона ( Huntington) , а также дистония и гемибаллизм. Патологические изменения в специфических областях базальных ганглиев значительно влияют на активность нейронов , распространяющуюся через все базально ганглионарные - таламокортикальные сети и активности нисходящих проекций на ствол мозга. Наиболее серьезные и грубые двигательные расстройства развиваются в результате дисфункции полосатого тела и субталамических ядер. Напротив, прерывание основных выходящих волокон из нейронов базальных ядер , внутреннего сегмента бледного шара , имеет небольшое значение или не отражается на двигательной активности. До сих пор причина столь разных нарушений двигательной активности остается сегодня неизвестной. вероятно, клиническая картина специфических двигательных расстройств определяется от комбинации изменений т степени нагрузки и паттернов , синхронизирующих необходимую для нее активность и в определенной степени зависит от индивидуальных особенностей моторных субнейронных сетей. Гипокинетические расстройства характеризуются нарушением инициации двигательной активности ( акинезия) , редукцией амплитуды и скорости свободных движений ( брадикинезия) , мышечной ригидностью ( усиливающейся резистентностью или сопротивлением при пассивных движениях ) и 4-6 Гц тремором покоя, а также согнутым положением тела. Гиперкинетические расстройства, напротив характеризуются насильственными движениями , такими как хорея ( случайные фрагментарные движения отдельных частей тела) , баллизмом ( движениями большой амплитуды , особенно в проксимальных отделах конченостей ), а также дистонией ( замедленные скручивающие движения и сохранением аномальной позы).
В основе паркинсонизма лежит дефицит дофамина в базальных ганглиях. Болезнь Паркинсона , впервые описанная в 1817 году поражает свыше миллиона людей в Северной Америке. В дополнение к основным симптомам - акинезии, брадикинезии , мышечной ригидности и тремору , другие моторные нарушения включают в себя: нарушение походки, согнутую позу, редцированную экспрессию лицевых мышц , редкое мигание , небольшие нарушение письма. Другая клиническая особенность паркинсонизма - уменьшение автоматической двигательной активности. Этиология болезни Паркинсона остается до конца неясной, но, вероятно, она связана с комбинацией наследственной предрасположенности и влиянием факторов внешней среды. Хроническая интоксикация пестицидами , проживание в сельской местности и потребление больших объемов воды способствуют возникновению данной болезни. Вероятно, митохондриальные токсины могут отрицательно влиять на активность дофаминергических нейронов нарушая их энергетический метаболизм. К факторам препятствующим развитию болезни Паркинсона следует отнести курение и частое потребление коффеина. Простые генетические мутации могут также предрасполагать к возникновению паркинсонизма.
Нарушения немоторных базально ганглионарн- таламо - кортикальных нейронных сетей могут обусловливать развитие когнитивных и бихевиоральных нарушений, сопровождающих двигательные расстройства , а также такого психического расстройства как обсессивно - компульсивное, синдрома Туретта и депрессии. В экспериментальных исследованиях на животных введение микроинъекций антагонистов GABA рецепторов ( бикукуллин ) в моторные, лимбические и ассоциативные волокна внешнего паллидарного сегмента у приматов способствует развитию у приматов различных нейроповеденческих синдромов , возникающих вследствидисфункции базально ганглионарных - таламокортикальных циклов. Инъекции в лимбическую часть внешнего сегмента бледного шара индуцирует стереотипные движения , в то время , как инъекции в ассоциативную часть индуцируют гиперактивность. Аномальные движения появляются только в случае введения бикукуллина в моторную область. Результаты этих исследований говорят о наличии в базальных ганглиях доменов связанных с поведением пациентов и роли этих структур в аномальной моторной и не моторной активности.
Поражение дорсолатерального префронтального кортекса или субкортикальной части префронтальных нейронных сетей сопровождается нарушениями когнитивного функционирования или исполнительного функционирования , в частности, в то время как повреждение латеральной орбитофронтальной нейронной сети ( круга) асоциируется с отсутствием эмпатии , эмоциональной лабильностью , раздражительностью и слабым ответом на социальные стимулы. Одним из хорошо изученных психических расстройств, возникновение которого связано с патологией не моторных нейронных кругов является обсессивно - компульсивное расстройство. Стереотипное поведение ( ригидные бихевиоральные паттерны) и компульсии являются характерными проявлениями этой психической болезни, вероятно, в генезе которой принимает участие дисфункция процедурального обучения ( procedural learning). Функциональное нейровизуализационное исследование пациентов с обсессивно - компульсивным расстройством демонстрирует нарушение активности в базальныг ганглиях - таламокортикальных лимбических нейронных связях , которые направляют свои волокна в орбиофронтальный и передний цингулярный ( поясной) кортекс. Наиболее значительные изменения в вентральном стриатуме , особенно в прилежащих ядрах и вентромедиальном хвостатом ядре и среднем мозге здесь играют активную роль. Позитивные результаты нейрохирургического лечения , направленные на лимбические нейронные связи , такие как разрез или стимуляция переднего лимба внутренней капсулы и вентрального стриатума или перерезка волокон , выходящих из орбитофронтального или переднего цингулярного кортекса , вероятно, обяъсняет роль этих структур в генезе обсессивно - компульсивного расстройства. Синдром Туретта , при котором обсессивно - компульсивные расстройства связаны с тиками и вокализмами также характеризуется отклонениями в лимбических нейронных связях ( кругах). Препараты, блокирующие рецепторы дофамина подавляют тики вызванные нарушением базальных ганглиев. Дополнительные изменения в активности некоторых областей коры мозга, связанными с особенно моторным кортексом и с моторными функциями и добавочными моторными областями также влияют на двигательные расстройства. Хроническая стимуляция моторных и лимбических связей между нейронами на паллидарном и таламическом уровнях сегодня применяется для лечения резистетных к терапии вариантов синдрома Туретта.
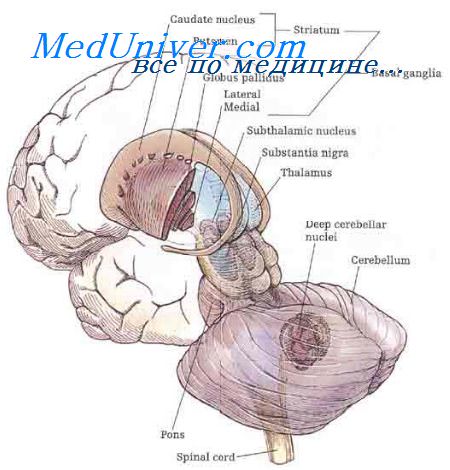
К экстрапирамидной системе анатомически относятся те проводящие пути и центры, которые не принадлежат к кортико-спинальной или кортикобульбарной системе, но оказывают свое влияние на характер движений. Они представляют собой системы волокон:
• проводящие импульсы от прецентральных, а также височных и теменных областей головного мозга к мосту и мозжечку (кортико-мосто-мозжечковый путь), а также
• проводящие импульсы от коры головного мозга к базальным ганглиям (полосатое тело, хвостатое ядро и скорлупа, красное ядро, черная субстанция и ретикулярная формация в стволе мозга) и, наконец,
• пути, которые идут от нейронов вышеназванных ядерных областей через промежуточные нейроны в спинной мозг (тектоспинальный, вестибулоспинальный и ретикулоспинальный пути);
• не всегда в анатомическом, но в функциональном смысле к экстрапирамидной системе относится также ряд пучков волокон, связывающих базальные ганглии друг с другом и с корой мозга, которые вместе с таламусом и мозжечком являются частью комплексной системы регуляции.
Эта система выполняет ряд важных функций в осуществлении движений:
• она участвует в регуляции мышечного тонуса;
• она регулирует автоматизм и оптимальную координацию многих движений;
• она обеспечивает гармоничное и экономичное выполнение движения, оптимальную последовательность его отдельных компонентов. В этом основную роль играют два вещества-нейротрансмиттера, которые действуют в разных отделах системы: дофамин и ацетилхолин.
Для синдромов поражения базальных ганглиев и экстрапирамидных синдромов в целом характерны следующие признаки:
• Нейроны бледного шара и черной субстанции понижают мышечный тонус, а при поражении этих структур наблюдается его повышение в виде ригидности. Нарушение функции хвостатого ядра, скорлупы или субталамического ядра Льюиса (а также мозжечка) вызывает понижение тонуса.
• Двигательные нарушения могут носить различный характер:
- гипокинезия и расстройство первичных двигательных автоматизмов с уменьшением содружественных движений наблюдаются при уменьшении активирующего влияния бледного шара или черной субстанции на двигательную активность (гипокинетико-гипертонический синдром); - непроизвольные движения различной степени выраженности, обычно в сочетании с мышечной гипотонией, развиваются при нарушении функции неостриатума (скорлупы и хвостатого ядра). При рано развившихся нарушениях (в пре- и раннем постнатальном периоде) преобладает атетоз, при более поздних — хореоатетоидные движения.
Баллизм наблюдается при наличии очага в субтаталамическом ядре и наружных отделах бледного шара. Дистонические синдромы и торсионная дистония развиваются прежде всего при нарушении функции скорлупы или связей между центральными ядрами таламуса и скорлупой. Психические нарушения могут (в зависимости от этиологии заболевания) сопровождать некоторые синдромы поражения базальных ганглиев: дисфория, депрессия, навязчивые состояния и навязчивые мысли при синдроме паркинсонизма, раздражительность и эмоциональная лабильность при малой хорее, деменция при хорее Гентингтона.
Среди многообразных этиологических факторов следует упомянуть:
• дегенеративные заболевания (например, болезнь Паркинсона), часть из которых представляет собой наследственные болезни (например, хорея Гентингтона) в качестве наиболее частой причины;
• нарушения обмена веществ, как. например, гепатолентикулярная дегенерация (болезнь Вильсона);
• генетически обусловленные дефекты ферментов, например системная дегенерация при оливопонтоцеребеллярной атрофии с синдромом паркинсонизма в рамках недостаточности глутаматдегидрогеназы;
• эндокринные расстройства, например обратимый синдром паркинсонизма при гипопаратиреозе;
• воспалительные заболевания, например ревматическая малая хорея (хорея Сиденхема);
• токсическое воздействие, например синдром паркинсонизма при отравлении марганцем или приеме производных хлорпромазина;
• аноксическое (сосудистое) повреждение, например торсионная дистония после родовой травмы;
• иногда наблюдаются опухоли или другие объемные процессы с локализацией в области субталамического ядра Льюиса, которые могут быть причиной гемибаллизма.
При одном классе экстрапирамидных заболеваний, представленном болезнью Паркинсона, основной дефект проявляется акинезией или гипокинезией, что характеризует неспособность пациента совершать быстрые движения в конечностях. Снижение двигательной активности (гипокинезия, или обеднение движений) распространяется и на мелкие автоматизированные движения, постоянно присутствующие у здорового человека (пациент, страдающий паркинсонизмом, сидит неподвижно, лицо маскообразное). Произвольные движения и движения по заданию выполняются с некоторой задержкой (замедление времени реакции) и замедленны (брадикинезия). В основе по-видимому, лежит нарушение быстрых (баллистических) движений. Для совершения произвольного движения необходима неоднократная повторная активация мышц-агонистов. Выполнение чередующихся движений затруднено. Брадикинезия, как правило, сопровождается ригидностью, но не вызвана ею.
Ригидность — второй компонент синдрома паркинсонизма В отличие от спастичности, мышечный тонус повышен по пластическому типу в виде равномерного сопротивления, возникающего в мышцах-агонистах и антагонистах в начале произвольного или пассивного движения и сохраняющегося на всем его протяжении. Снижение мышечной силы и изменение сухожильных рефлексов выражены незначительно.

Вышеперечисленные компоненты синдрома паркинсонизма — наиболее частые проявления поражения нигростриальной системы; однако более обширные очаги поражения, вовлекающие полосатое тело (стриатум), паллидум (бледный шар) и черную субстанцию, могут привести к появлению изолированной ригидности.
Непроизвольные движения — хорея, баллизм, атетоз и дистония — другие характерные симптомы заболеваний базальных ганглиев.
Хорея — аритмичные непроизвольные быстрые беспорядочные движения, вовлекающие пальцы руки, кисть, всю конечность или другие части тела. Гримасничанье, респираторные звуки представляют другие проявления хореи. В промежутках между непроизвольными движениями тонус в пораженных конечностях снижен. Хорея может быть ограничена одной стороной тела (гемихорея); если в движения вовлекаются проксимальные мышцы конечностей и они необычно интенсивны и размашисты, расстройство расценивается как гемибаллизм. В отличие от других непроизвольных движений, для гемибаллизма установлена определенная локализация поражения — противоположное субталамическое ядро.
Хореический гиперкинез — основное проявление хореи Сиденгама (малой хореи) и хореи беременных, которые, вероятно, представляют собой заболевания иммунной природы, связанные с ревматизмом, а также хореи Гентингтона, однако при ней движения имеют характер хореоатетоза. Гемихореоатетоз может появляться после гемиплегии при частичном восстановлении. Избыточный прием леводофы пациентами, страдающими паркинсонизмом, приводит к появлению ограниченного или генерализованного хореоатетоза, который также служит наиболее частым проявлением поздней дискинезии вследствие длительного приема нейролептиков. Хореоатетоз также наблюдается при многих наследственных метаболических заболеваниях.
Атетоз — относительно медленные червеобразные непроизвольные движения, сменяющиеся одно другим. Конечности поочередно принимают положение сгибание-супинация и разгибание-пронация. В промежутке между непроизвольными движениями тонус мышц конечностей может быть повышен по типу спастичности или ригидности, что определяется локализацией основного заболевания, однако чаще наблюдается гипотония. Одновременное сокращение мышц агонистов и антагонистов препятствует выполнению точных целенаправленных движений. При попытке выполнения движения, требующего участия группы мьшщ-агонистов, возникает одновременное сокращение расположенных рядом мышц (интенционный спазм).
Атетоз может быть генерализованным, что наблюдается при болезни Гентингтона, двойном атетозе (вследствие перинатальной гипоксии), хронической печеночной энцефалопатии, отравлении лекарственными препаратами (фенотиазиды, галоперидол, леводофа), различных дегенеративных заболеваниях базальных ганглиев, а также ограниченным определенной группой шейных или краниальных мышц, что наблюдается в случае идиопатической оромандибулярной дистонии, поздней дискинезии и спастической кривошее. Описана редко встречающаяся семейная форма пароксизмального хореоатетоза. Атетоз и хорея усиливаются при усталости и волнении и уменьшаются в покое.
Дистопия или торсионный спазм проявляются возникновением патологических поз, как правило, вследствие атетоидных движений в одной или нескольких конечностях, туловище, плечевом и тазовом поясе, кисти и стопе. Дистонические позы также возникают и без атетоза, они могут быть вначале периодическими или обратимыми, но позже становятся стойкими. Для Дистонии характерно одновременное сокращение мышц агонистов и антагонистов, что вызывает патологическую позу в вовлеченной части тела. Как и хореоатетоз, дистония служит проявлением многих наследственных дегенеративных заболеваний, а также осложнением при кратковременном или длительном приеме некоторых лекарственных препаратов (фенотиазиды, галоперидол) либо является локальной формой экстрапирамидного заболевания, вовлекающего мышцы лица, рта, нижней челюсти, языка, шеи или кистей рук (подробнее см. в полном варианте этой книги на английском языке).
Хореические, атетоидные и диатонические гиперкинезы так часто сочетаются, что, возможно, их разграничение не столь важно. В некоторых случаях ситуация еще более усложняется при сочетании этих гиперкинезов с тремором, миоклониями и атаксией. В этой связи некоторые исследователи избегают сомнительных классификаций гиперкинезов и называют все виды последних дискинезиями. Предполагаемая локализация поражения, лежащая в основе экстрапирамидных двигательных нарушений, представлена в таблице.
Рубрика МКБ-10: G23.8
Содержание
- 1 Определение и общие сведения
- 2 Этиология и патогенез
- 3 Клинические проявления
- 4 Другие уточненные дегенеративные болезни базальных ганглиев: Диагностика
- 5 Дифференциальный диагноз
- 6 Другие уточненные дегенеративные болезни базальных ганглиев: Лечение
- 7 Профилактика
- 8 Прочее
- 9 Источники (ссылки)
- 10 Дополнительная литература (рекомендуемая)
- 11 Действующие вещества
Двусторонний стриопаллидодентатный кальциноз
Синонимы: феррокальциноз сосудов головного мозга, идиопатический кальциноз базальных ганглиев.
Идиопатический кальциноз базальных ганглиев, его также ошибочно называют болезнью Фара, характеризуется накоплением отложений кальция в различных областях мозга, особенно в базальных ганглиях и зубчатом ядре, часто ассоциируется с нейродегенерацией.
Распространенность неизвестна, менее 200 случаев заболевания были зарегистрированы на сегодняшний день. Чаще встречается у мужчин (соотношение мужчин и женщин 2: 1).
Патология носит семейный или спорадический характеро. Известно более 30 семей с наследственной формой патологии, которая наследуется аутосомно-доминантно.
Идиопатический кальциноз базальных ганглиев является генетически гетерогенной патологией. С возникновением заболевания связывают локусы генов IBGC1 (14q11.2–21.3.), IBGC2 (2q37), IBGC3 (8p21.1-q11.13) и IBGC4 (5q32). Мутация гена SLC20A2 (локус IBGC3) была обнаружена у 218 пациентов из 29 семей. Еще один локус был идентифицирован в 2013 году и связан с геном PDGFRB, кодирующем тромбоцитарный бета фактор роста (PDGFB).
Идиопатический кальциноз базальных ганглиев может протекать бессимптомно. Симптоматические формы, как правило, манифестируют на четвертом десятилетии жизни, хотя кальцификации могут быть найдены во втором десятилетии. У пациентов наблюдаются двигательные расстройства, в том числе паркинсонизм, хорея, тремор, дистония, атетоз, орофациальная дискинезия и атаксия. Кроме того пациенты демонстрируют психоневрологические расстройства - затруднение концентрации внимание, нарушения памяти и/или изменения личности и поведения, а также признаки деменции. Часто первыми симптомами заболевания являются моторная неуклюжесть, утомляемость, неустойчивая походка, медленная или невнятная речь, дисфагия, непроизвольные движения или судороги. Может возникнуть недержание мочи .
Диагноз основывается на данных КТ или МРТ, демонстрирующих двусторонние, почти симметричные кальцификации одной или нескольких из следующих областей: базальных ганглиев, зубчатых ядер, таламуса и коры головного мозга. Электроэнцефалограмма, исследование нервной проводимости, зрительные вызванные потенциалы и слуховые вызванные потенциалы, как правило, нормальные или с незначительными отклонениями.
Дифференциальный диагноз включает гипопаратиреоз и псевдогипопаратиреодизм, синдром Кенни-Каффи 1-го типа, нейродегенерации с накоплением железа, синдром Коккейна и синдром Айкарди-Гутьерреса.
Специфического лечения на сегодняшний день нет. Лечение основано на купировании симптомов тревоги, депрессии, обсессивно-компульсивное поведения и дистонии.
Обызвествление базальных ядер и зубчатого ядра может сопутствовать некоторым другим состояниям (см. табл. 15.7), нередко оно выявляется у пожилых и может быть одной из причин легких двигательных нарушений, часто встречающихся в этой возрастной группе. При выраженном обызвествлении бывают тяжелые прогрессирующие двигательные расстройства в виде паркинсонизма или хореоатетоза.
В случае гипокальциемии (при послеоперационном или идиопатическом гипопаратиреозе либо при псевдогипопаратиреозе) прогрессирование двигательных расстройств можно остановить, если нормализовать концентрацию кальция в крови с помощью витамина D (50 000—100 000 МЕ/сут) и препаратов кальция. Для предупреждения передозировки витамина D необходимо регулярное биохимическое исследование крови. Регресс двигательных нарушений наблюдается редко, за исключением, возможно, случаев идиопатического гипопаратиреоза. Реже обызвествление возникает при гиперпаратиреозе и псевдопсевдогипопаратиреозе (нормокальциемическом псевдогипопаратиреозе).
Болезнь Гентингтона- наследственное медленно прогрессирующее заболевание нервной системы с аутосомно-доминантным типом наследования, характеризующееся хореическими гиперкинезами, психическими нарушениями и прогрессирующей деменцией. Частота встречаемости в популяции колеблется и составляет в среднем 3-7 на 100 000.
Историческая справка.Дж. Гентингтон был потомственным врачом. Под наблюдением его дедушки находилось несколько пациентов с наследственной формой хореи. Восьмилетний Джордж впервые увидел и зарисовал их движения. В 1872 г. Гентингтон впервые охарактеризовал это заболевание, впоследствии названное в его честь.
Молекулярная генетика и патогенез.Ген болезни Гентингтона картирован на хромосоме 4p16.3. Он кодирует белок гентингтин. Причиной болезни Гентингтона является увеличение числа тринуклеотидных цитозин-аденин-гуанин (САG)-повторов, расположенных в первом экзоне гена. В генах здоровых людей содержится от 10 до 35 повторов. При хорее Гентингтона наблюдается увеличение их числа (от 36 до 121). После того, как число тринуклеотидных повторов превысит 36, наблюдается накопление зоны повторов в последующих поколениях, что коррелирует с увеличением тяжести заболевания. Это явление получило название антиципации, и болезнь Гентингтона является лучшим его примером: чем раньше проявилось заболевание в ряду поколений, тем тяжелее оно протекает. Триплет CAG кодирует аминокислоту глутамин, поэтому в белке образуется удлиненный полиглутаминовый участок, который при- водит к апоптозу. При болезни Гентингтона также нарушается функция митохондрий в нейронах полосатого тела. Эти изменения, вероятно, обусловлены накоплением свободных перекисных радикалов.
Патоморфология.При аутопсии головного мозга при болезни Гентингтона обнаруживают атрофию и глиоз хвостатых ядер и скорлупы (рис. 1). Уменьшено количество нейронов в бледном шаре, в коре лобных долей и субкортикальных отделах полушарий. Специфических гистологических маркёров не описано. В непо- врежденных нейронах и астроцитах накапливается липофусцин, в клетках бледного шара - железо, в периваскулярном пространстве - сидерофаги. В основном повреждаются нейроны хвостатых ядер, ответственные за секрецию тормозящего нейромедиатора - γ-аминомасляной кислоты.
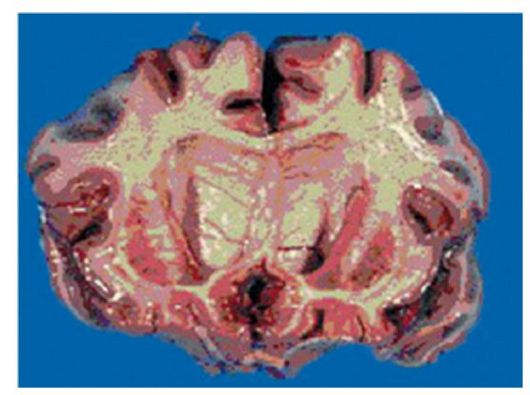
Рис. 1Атрофия головного мозга, преимущественно хвостатого ядра, при болезни Гентингтона (макропрепарат)Большие пирамидные клетки III, V и VI слоев коры большого мозга сморщиваются, приобретая неправильную форму. В начале болезни гибель клеток коры компенсируется за счет активного ветвления дендритов оставшихся пирамидных клеток.
Клинические проявления.Заболевание начинается в любом возрасте, чаще - в период с 20 до 60 лет (в среднем - в 40 лет). На ювенильную форму приходится около 10% всех случаев хореи Гентингтона. Самый ранний дебют заболевания описан в 3 года. В начальной стадии заболевания непроизвольные движения в виде хореи возникают утром или при нервном напряжении. Хореические гиперкинезы в лицевой мускулатуре проявляются выразительными гримасами с высовыванием языка, подергиванием щек, поочередным подниманием и нахмуриванием бровей. Иногда отмечаются эпизоды шумного, глубокого дыхания. Хорея в руках выглядит как быстрое сгибание и разгибание пальцев, в ногах - как поочередное скрещивание и разведение ног, сгибание и разгибание пальцев стоп. Наряду с хореей в мышцах туловища и проксимальных отделах конечностей можно отметить атетоз. Гиперкинезы обычно симметричны, усиливаются при физической нагрузке или волнении и прекращаются во сне. По мере развития болезни они усиливаются, появляется грубая дистония, переходящая в ригидность. Иногда заболевание начинается с дистонии: больные не могут длительно находиться в одной позе, отмечается торсия шеи, туловища и конечностей. При ювенильной форме в 50% случаев начальными симптомами являются брадикинезия, ригидность и паркинсонический тремор.Судороги у взрослых с болезнью Гентингтона бывают редко, а у детей встречаются в 30-50% случаев. Наблюдаются различные типы приступов: фокальные, генерализованные тонико-клонические, абсансы, диалептические, миоклонические, обычно резистентные к противосудорожным препаратам. Изменения на ЭЭГ характеризуются генерализованной эпилептической активностью с частотой 2- 2,5 Гц и нерегулярными пик-волнами. У больных прогрессируют расстройства речевых функций. На начальных стадиях хореи Гентингтона возникают нарушения, связанные со звукопроизношением (дизартрия). Постепенно изменяются скорость и ритм речи, она становится медленной и невнятной. Нарушения глотания обычно появляются в терминальной стадии. Частой причиной смерти является аспирационный синдром. У 90% детей выявляют повышение сухожильных рефлексов и спастический гипертонус. Аксиальные рефлексы (хоботковый, сосательный, дистансоральный), как правило, возникают при грубых интеллектуальных нарушениях. Глазодвигательные нарушения встречаются у большинства пациентов. Больные не могут плавно и точно следить за предметом, часто моргают. Характерен нистагм. Часто болезнь Гентингтона в детском возрасте начинается с изменений поведения: снижаются успеваемость в школе и концентрация внимания, замедляется мышление, нарушается кратковременная память, появляется неусидчивость. Редко в подростковом возрасте заболевание дебютирует с психозов, шизотипического расстройства. Для начальной стадии характерны снижение настроения (депрессия), тревога, раздражительность, эмоциональная лабильность, апатия. Возникают суицидальные мысли. Течение заболевания у детей характеризуется быстрым прогрессированием, что связано с феноменом антиципации.
Диагностика.Диагноз подтверждается при молекулярно-генетическом анализе. С помощью полимеразной цепной реакции определяют число САG-повторов в пораженном гене. При взрослой форме заболевания число повторов превышает 36, при ювенильной - 50. На МРТ головного мозга видна атрофия головок хвостатых ядер, в меньшей степени - бледных шаров и гипоталамуса, лобных отделов коры. Однофотонная эмиссионная компьютерная томография (SPECT) выявляет низкий метаболизм глюкозы в хвостатых ядрах еще на доклинической стадии.
Дифференциальный диагнозпроводят с другими заболеваниями детского возраста, проявляющимися хореей: доброкачественной непрогрессирующей семейной хореей, идиопатической торсионной дистонией, болезнью Галлервордена-Шпатца, болезнью Вильсона- Коновалова, ювенильной формой болезни Паркинсона, нейроакантоцитозом. Пренатальная диагностика проводится молекулярно-генетическим методом.
Лечение.В настоящее время эффективного лечения не разработано, проводят симптоматическую терапию. Для уменьшения выраженности хореи показаны нейролептики. При ригидности назначают препараты леводопа, бромокриптин, амантадин, при возникновении судорог - антиэпилептическую терапию.
В основе заболевания лежит нарушение метаболизма меди. Ион меди входит в состав ферментов дыхательной цепи (цитохромоксидазы и лизилоксидазы). Ежедневно человек употребляет с пищей от 1 до 5 мг меди, из которых усваивается около 40%. Всосавшиеся в проксимальных отделах ЖКТ ионы меди образуют прочное соединение с металлопротеином, транспортируются в клетки, участвуют во внутриклеточном обмене и экскретируются. При болезни Вильсона- Коновалова нарушается выведение меди из печени в составе церуллоплазмина. Медь накапливается в гепатоцитах, развивается гепатоз, а в дальнейшем - нодулярный цирроз печени. Непосредственное токсическое воздействие меди вызывает гемолитическую анемию.Свободно циркулирующая медь откладывается в органах и тканях, в первую очередь в головном мозге и роговице. Формируются патологические изменения в базальных ядрах и кольцо Кайзера-Флейшера в роговице. Хроническая интоксикация приводит к поражению ЦНС. Летальный исход наступает от печеночной комы.
Брюшная форма- тяжелое заболевание печени, приводящее к смерти до появления симптомов со стороны нервной системы; заболевают дети дошкольного возраста.
Ригидно-аритмо-гиперкинетическая, или ранняя, формаотличается быстрым течением (2-3 года), начинается также в детском возрасте. В клинической картине заболевания преобладают мышечная ригидность,приводящая к контрактурам, бедность и замедленность движений, хореоатетоз или дистонические гиперкинезы; лицо амимично, часто искажено застывшей гримасой. Обычны расстройства речи (дизартрия) и глотания (дисфагия), судорожные смех и плач, нередки судороги, аффективные расстройства и умеренное снижение интеллекта.
Дрожательно-ригидная формавстречается чаще других. Начинается в юношеском возрасте, течет несколько медленнее (в среднем 5-6 лет), порой сопровождаясь ремиссиями и внезапными ухудшениями. Типичны грубая ригидность и ритмичный тремор (2-8 дрожаний в 1 с), который резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает; захватывает конечности, голову и туловище. Иногда к тремору присоединяются атетоз и хорея, наблюдаются также дисфагия и дизартрия.
Дрожательная форманачинается в возрасте 20-30 лет, течет довольно медленно (10 лет и больше); в клинике преобладает тремор, ригидность появляется в конце болезни. Нередки гипотония, амимия, медленная, монотонная речь (брадилалия), брадикинезия, изменения психики, аффективные вспышки. Наблюдаются эпилептические приступы
Экстрапирамидно-корковая формавстречается реже других форм, длится 6-8 лет; начинается как одна из вышеописанных форм. Типичные экстрапирамидные нарушения в дальнейшем осложняются остро развивающимися парезами, судорогами и слабоумием, которые связаны с образованием обширных очагов в коре больших полушарий.
Читайте также:
