
Диагностика миелопатии и бас


Боковой (латеральный) амиотрофический склероз (БАС) (также известен как болезнь моторных нейронов, Мотонейронная болезнь, болезнь Шарко, в англоязычных странах — болезнь Лу Герига) — прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних (моторная кора головного мозга), так и нижних (передние рога спинного мозга и ядра черепно-мозговых нервов) двигательных нейронов, что приводит к параличам и последующей атрофии мышц.
Болезнь известна не так давно. Впервые описана Жан-Мартеном Шарко в 1869г. По статистике выявляется у 2-5 человек на 100 000 населения в год, что говорит о том, что данная патология относительно редко встречается. Всего в мире насчитывается около 70 тысяч больных боковым амиотрофическим склерозом. Обычно заболевание заявляет о себе у людей старше 50 лет.
Совсем недавно было высказано мнение, что случаи бокового амиотрофического склероза чаще регистрируются у высокоинтеллектуальных людей, профессионалов в своем деле, а также у спортсменов-атлетов, которые на протяжении всей жизни отличались крепким здоровьем.
В 90% случаев БАС носит спорадический, а в 10% - семейный или наследственный характер как с аутосомно-доминантным (преимущественно), так и с аутосомно-рецессивным типами наследования. Клинические и патоморфологические характеристики семейного и спорадического БАС практически идентичны.
Точная этиология БАС неизвестна.
Сущность болезни заключается в дегенерации двигательных нейронов, т.е. под воздействием ряда причин запускается процесс разрушения нервных клеток, ответственных за сокращения мышц. Этот процесс затрагивает нейроны коры больших полушарий, ядер головного мозга и нейроны передних рогов спинного мозга. Двигательные нейроны погибают, а их функции никто больше не выполняет. Нервные импульсы к мышечным клеткам больше не поступают. И мышцы слабеют, развиваются парезы и параличи, атрофия мышечной ткани.
Предполагается, что роль других факторов в развитии БАС также заключается в запуске свободнорадикального окисления.
Классификация БАС, формы:
- пояснично-крестцовая;
- шейно-грудная;
- бульбарная: при поражении периферического мотонейрона в стволе головного мозга;
- высокая: при поражении центрального мотонейрона.
Общими симптомами, характерными для любой из форм бокового амиотрофического склероза, являются:
- сугубо двигательные нарушения;
- отсутствие чувствительных расстройств;
- отсутствие расстройств со стороны органов мочеиспускания и дефекации;
- неуклонное прогрессирование болезни с захватом новых мышечных массивов вплоть до полной обездвиженности;
- наличие периодических болезненных судорог в пораженных частях тела, их называют крампи.
Начальные проявления заболевания:
•слабость в дистальных отделах рук, неловкость при выполнении тонких движений пальцами, похудание в кистях и фасцикуляции (мышечные подергивания)
•реже заболевание дебютирует слабостью в проксимальных отделах рук и плечевом поясе, атрофиями в мышцах ног в сочетании с нижним спастическим парапарезом
•возможно также начало заболевания с бульбарных расстройств – дизартрии и дисфагии (25% случаев)
•крампи (болезненные сокращения, спазмы мышц), нередко генерализованные, встречаются практически у всех больных БАС, и нередко являются первым признаком заболевания
Для БАС в большинстве случаев характерна асимметричность симптоматики.
При этой форме заболевания возможно два варианта:
Также может дебютировать двумя способами:
- При этой форме заболевания первыми симптомами при поражении периферического мотонейрона в стволе мозга становятся расстройства артикуляции, поперхивание при приеме пищи, гнусавость голоса, атрофия и фасцикуляции языка. Движения языка затруднены. Если поражен и центральный мотонейрон, то к этим симптомам присоединяются и повышение глоточного и нижнечелюстного рефлексов, насильственный смех и плач. Повышается рвотный рефлекс.
В руках по мере прогрессирования болезни формируется парез с атрофическими изменениями, повышением рефлексов, повышением тонуса и патологическими стопными признаками. Аналогичные изменения возникают и в ногах, но несколько позже.
Это разновидность бокового амиотрофического склероза, когда заболевание протекает с преимущественным поражением центрального мотонейрона. При этом во всех мышцах туловища и конечностей формируются парезы с повышением мышечного тонуса, патологическими симптомами.
Бульбарная и высокая формы БАС являются прогностически неблагоприятными. Больные с таким началом заболевания имеют меньшую продолжительность жизни по сравнению с шейно-грудной и пояснично-крестцовой формами. Какими бы ни были первые проявления заболевания, оно неуклонно прогрессируют.
Парезы в различных конечностях приводят к нарушению способности самостоятельно передвигаться, обслуживать себя. Вовлечение в процесс дыхательной мускулатуры приводит вначале к появлению одышки при физической нагрузке, затем одышка беспокоит уже в покое, появляются эпизоды острой нехватки воздуха. В терминальных стадиях самостоятельное дыхание просто невозможно, больным требуется постоянная искусственная вентиляция легких.
Продолжительность жизни больного БАС составляет по разным данным от 2 до 12 лет, однако более 90% больных умирают в течение 5 лет от момента постановки диагноза. В терминальную стадию болезни больные полностью прикованы к постели, дыхание поддерживается с помощью аппарата искусственной вентиляции легких. Причиной гибели таких больных может стать остановка дыхания, присоединение осложнений в виде пневмонии, тромбоэмболии, инфицирования пролежней с генерализацией инфекции.
Среди параклинических исследований наиболее существенное диагностическое значение имеет электромиография. Выявляется распространенное поражение клеток передних рогов (даже в клинически сохранных мышцах) с фибрилляциями, фасцикуляциями, позитивными волнами, изменениями потенциалов двигательных единиц (увеличивается их амплитуда и длительность) при нормальной скорости проведения возбуждения по волокнам чувствительных нервов. Содержание КФК в плазме может быть незначительно повышено
Боковой амиотрофический склероз нужно заподозрить:
•при развитии слабости и атрофий, а возможно и фасцикуляций (мышечных подергиваний) в мышцах кисти
•при похудания мышц тенара одной из кистей с развитием слабости аддукции (приведения) и оппозиции большого пальца (обычно асимметрично)
•при этом наблюдается затруднение при схватывании большим и указательным пальцами, затруднения при подбирании мелких предметов, при застегивании пуговиц, при письме
•при развитии слабости в проксимальных отделах рук и плечевом поясе, атрофий в мышцах ног в сочетании с нижним спастическим парапарезом
•при развитии у пациента дизартрии (нарушений речи) и дисфагии (нарушений глотания)
•при появлении у пациента крампи (болезненных мышечных сокращений)
Диагностические критерии БАС:
- Симптомы поражения нижнего моторного нейрона (включая ЭМГ-подтверждение в клинически сохранных мышцах).
- Симптомы поражения верхнего моторного нейрона
- Прогрессирующее течение
Критерии исключения БАС
Для диагностики бокового амиотрофического склероза необходимо отсутствие:
•сенсорных расстройств, в первую очередь выпадений чувствительности (возможны парестезии и боли)
•тазовых расстройств - нарушений мочеиспускания и дефекации (их присоединение возможно на конечных стадиях заболевания)
•зрительных нарушений
•вегетативных нарушений
•болезни Паркинсона
•деменции альцгеймеровского типа
•синдромов, похожих на БАС
Критерии подтверждения БАС:
Диагноз БАС подьверждается:
- Фасцикуляциями в одной и более областях
- ЭМГ-признаки нейронопатии
- Нормальной скоростью проведения возбуждения по моторным и сенсорным волокнам (дитсальные моторные латенции могут быть увеличенными)
- Отсутствием блока проведения
Дифференциальный диагноз БАС (синдромы похожие на БАС):
•Спондилогенная шейная миелопатия.
•Опухоли краниовертебральной области и спинного мозга.
•Краниовертебральные аномалии.
•Сирингомиелия.
•Подострая комбинированная дегенерация спинного мозга при недостаточности витамина В12.
•Семейный спастический парапарез Штрюмпеля.
•Прогрессирующие спинальные амиотрофии.
•Постполиомиелитический синдром.
•Интоксикации свинцом, ртутью, марганцем.
•Недостаточность гексозаминидазы типа А у взрослых при ганглиозидозе GM2.
•Диабетическая амиотрофия.
•Мультифокальная моторная невропатия с блоками проведения.
•Болезнь Крейцтфельдта-Якоба.
•Паранеопластический синдром, в частности при лимфогранулематозе и злокачественной лимфоме.
•Синдром БАС при парапротеинемии.
•Аксональная нейропатия при болезни Лайма (Лайм-боррелиозе).
•Синдром Гийена-Барре.
•Миастения.
•Рассеянный склероз
•Эндокринопатии (тиреотоксикоз, гиперпаратиреоз, диабетическая амиотрофия).
•Доброкачественные фасцикуляции, т.е. фасцикуляции, продолжающиеся годами без признаков поражения двигательной системы.
•Нейроинфекции (полиомиелит, бруцеллез, эпидемический энцефалит, клещевой энцефалит, нейросифилис, болезнь Лайма).
•Первичный боковой склероз.
Диагностические исследования при синдроме БАС.
Для уточнения диагноза и проведения дифференциального диагноза при синдроме БАС рекомендутся следующее обследование больного:
Анализ крови (СОЭ, гематологические и биохимические исследования)
Рентгенография органов грудной клетки
Исследование функций щитовидной железы
Определение содержания витамина В12 и фолиевой кислоты в крови
Креатинкиназа в сыворотке
МРТ головного мозга и при необходимости, спинного мозга
Эффективного лечения заболевания не существует. Единственный препарат, ингибитор высвобождения глутамата рилузол (Рилутек), отодвигает летальный исход на 2 – 4 месяца. Его назначают по 50 мг два раза в день.
Основу лечения составляет симптоматическая терапия:
•Физическая активность. Пациент должен по мере своих возможностей поддерживать физическую активность По мере прогрессирования заболевания возникает необходимость в кресле-каталке и других специальных приспособлениях.
•Диета. Дисфагия создаёт опасность попадания пищи в дыхательные пути • Иногда возникает необходимость в питании через зонд или в гастростомии.
•Применение ортопедических приспособлений: шейного воротника, различных шин, устройств для захвата предметов.
•При крампи (болезненным мышечных спазмах): карбамазепин (Финлепсин, Тегретол) и/или витамин Е, а также препараты магния, верапамил (Изоптин).
•При спастичности: баклофен (Баклосан), Сирдалуд, а также клоназепам.
•При слюнотечении атропин, или гиосцин (Бускопан).
•При невозможности приема пищи вследствие нарушения глотания накладывают гастростому или вводят назогастральный зонд. Раннее проведение чрезкожной эндоскопической гастростомии продлевает жизнь пациентов в среднем на 6 месяцев.
•При болевых синдромах используют весь арсенал аналгетиков. В том числе на конечных стадиях наркотические аналгетики.
•Иногда некоторое временное улучшение приносят антихолинэстеразные препараты (неостигмина метилсульфат - прозерин).
•Церебролизин в высоких дозах (10-30 мл в/в капельно 10 дней повторными курсами). Существует ряд небольших исследований, показывающих нейропротективную эффективность церебролизина при БАС.
•Антидепрессанты: Серталин или Паксил или Амитриптилин (часть больных БАС предпочитает именно его как раз из-за побочных действий – он вызывает сухость во рту, соответственно уменьшает гиперсаливацию (слюнотечение), часто мучающую больных БАС).
•При появлении дыхательных нарушений: искусственная вентиляция легких в условиях стационаров, как правило, не проводится, но некоторые больные приобретают портативные приборы ИВЛ и проводят ИВЛ в домашних условиях.
•Ведутся разработки к применению гормона роста, нейротрофических факторов при БАС.
•Последнее время активно ведутся разработки лечения стволовыми клетками. Этот метод обещает быть перспективным, но все же пока находится на стадии научных экспериментов.
•Боковой амиотрофический склероз является фатальным заболеванием. Средняя продолжительность жизни больных БАС 3 – 5 лет, тем не менее, 30% больных живут 5 лет, а около 10 – 20% живут более 10 лет от начала заболевания.
•Неблагоприятные прогностические признаки – пожилой возраст и бульбарные нарушения (после появления последних больные живут не более 1 – 3 лет).
Общие сведения
Миелопатия спинного мозга — это тяжелый соматический синдром, обобщающий различные по этиологическому признаку поражения спинного мозга, сопутствующий многочисленным патологическим процессам и проявляющийся нейродегенеративными изменениями в отдельных спинномозговых сегментах имеющий, как правило, хроническое течение.
Миелопатия всегда возникает вследствие различных патологических нарушений в организме (осложнение дегенеративно-дистрофических заболеваний позвоночника, травм и опухолей позвоночного столба, патологий сосудистой системы, токсического воздействия, соматических заболеваний и инфекционных поражений).
В зависимости от этиологического фактора, т.е. от заболевания, ставшего предпосылкой развития миелопатии, при постановке диагноза указывается эта болезнь/патологический процесс, например, сосудистая, диабетическая, компрессионная, алкогольная, вертеброгенная, ВИЧ-ассоциированная миелопатия и др., то есть таким образом указывается на происхождение синдрома (природу поражения спинного мозга). Очевидно, что при разных формах миелопатии спинного мозга лечение будет существенно отличаться, поскольку необходимо воздействие на основную причину, которая вызвала соответствующие изменения. По МКБ-10 миелопатия кодируется G95.9 (Болезнь спинного мозга неуточненная).
Достоверно точной информации о частоте встречаемости в целом миелопатии нет. Существует лишь информация о некоторых наиболее распространенных причинах ее формирования. Так в США ежегодно происходит от 12 до 15 тысяч травм спинного мозга, а у 5%-10% пациентов со злокачественными опухолями существует высокая вероятность метастазов в эпидуральное пространство позвоночника, что является причиной более 25 тысяч случаев миелопатии в год.
Некоторые виды миелопатии являются относительно редкими (сосудистая миелопатия), другие (цервикальная спондилогенная миелопатия) встречается у почти 50% лиц мужского пола и 33% у женщин в возрасте после 60 лет, что обусловлено выраженностью дегенеративных изменений в структурах позвоночного столба и нарастание проблем со стороны сосудистой системы, характерных для людей пожилого возраста. Наиболее часто поражается шейный и поясничные отделы позвоночника и значительно реже встречается миелопатия грудного отдела позвоночника.
Патогенез
Патогенез развития миелопатии существенно различается в зависимости от заболевания, вызвавшего тот или иной вид миелопатии. Во множестве случаев патологические процессы, лежащие в основе развития заболевания, локализуются вне спинного мозга и рассмотреть их в пределах одной статьи не представляется возможным.
Классификация
В основу классификации положен этиологический признак, в соответствии с которым выделяются:
- Вертеброгенная (дискогенная, компрессионная, спондилогенная) — может быть обусловлена, как травмами позвоночника (посттравматическая), так дегенеративными изменениями в позвоночном столбе (смещение позвонков, остеохондроз, спондилез с выраженным разрастанием остеофитов, стеноз спинномозгового канала, грыжа межпозвоночного диска и др.).
- Дисциркуляторная (ишемическая) — сосудистая, атеросклеротическая, дисциркуляторная, развивается вследствие медленно прогрессирующей хронической недостаточности (ишемии) спинномозгового кровообращения.
- Инфекционная — развивается под воздействием патогенной микрофлоры (энтеровирусы, вирус герпеса, бледная трепонема) и часто является следствием септицемии, пиодермии, остеомиелита позвоночника, СПИДа, болезни Лайма и др.
- Миелопатии, вызванные различного рода интоксикациями и физическими воздействиями (токсические миелопатии; лучевая миелопатия).
- Метаболическая — обусловлена метаболическими расстройствами и осложнением эндокринных заболеваний.
- Демиелинизирующая. В ее основе лежат патологические процессы, вызывающие разрушение (демиелинизацию) миелиновой оболочки нейронов, что приводит к нарушению процессов передачи импульсов между нервными клетками спинного и головного мозга (рассеянный склероз, болезнь Бало, болезнь Канавана и др.).
По локализации патологического процесса выделяется:
- Миелопатии шейного отдела позвоночника (син. цервикальная миелопатия).
- Миелопатии грудного отдела позвоночника.
- Миелопатии поясничного отдела.
Причины
К основным причинам развития миелопатий относятся:
- Компрессия (сдавливание), возникающая в результате травм позвоночника со смещением позвоночных сегментов, спондилолистеза, спондилеза, первичной/метастатической опухоли спинного мозга, эпидурального абсцесса и гематомы, субдуральной эмпиемы, грыжи межпозвонкового диска, туберкулезного спондилита, подвывиха в атлантоаксиальном сочленении, вторичного спаечного процесса и др.
- Нарушение кровообращения в спинном мозге, обусловленное вышеперечисленными причинами, так и различного рода сосудистой патологией, которая формирует медленно прогрессирующую хроническую недостаточность кровоснабжения: атеросклероз, эмболии, тромбозы, аневризма, венозный застой, развивающимся вследствие сердечно-легочной/сердечной недостаточности, сдавление венозных сосудов на различных уровнях позвоночника.
- Воспалительные процессы с локализацией в спинном мозге, обусловленные патогенной микрофлорой, травмой или в силу других обстоятельств (спинальный арахноидит, туберкулез, болезнь Бехтерева, миелит и др.).
- Нарушение процессов метаболизма в организме (гипергликемия при сахарном диабете).
Несмотря на многообразие причин, основной предпосылкой формирования миелопатии принято считать прогрессирующий длительно протекающий остеохондроз (вертеброгенная, дискогенная, компрессионная, дегенеративная миелопатия).
Симптомы
Симптомы миелопатии варьируют в широком диапазоне в зависимости от причин заболевания, уровня поражения, тяжести состояния, характера патологического процесса (острый/хронический). К общим симптомам относятся:
- Постоянный/возникающий при движении интенсивный болевой синдром в спине ноющего/тупого характера.
- Онеменение верхних/нижних конечностях, слабость, нарушения мелкой моторики (при застегивании одежды, письме и др.).
- Снижение в различной степени температурной и болевой чувствительности, появление дисфункции тазовых органов (мочеиспускания).
- Развитие сочетанных спастических парезов и параличей, вызывающих нарушения походки.
Из всего многообразия видов миелопатий рассмотрим лишь несколько, наиболее часто встречающихся в тех или иных отделах позвоночника.
Шейная спондилогенная миелопатия относится к одной из частых причин дисфункции спинного мозга нетравматического характера у людей старшего возраста с развитием спастического тетра- и парапареза. Ведущим патофизиологическим механизмом этого заболевания является ишемия спинного мозга, обусловленная его компрессией с нарастающими дегенеративными процессами структур шейного отдела позвоночного столба (фото ниже).
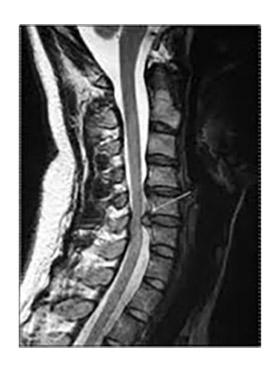
Симптомы отражают нарушения функции верхнего мотонейрона, поражение задних столбов спинного мозга и пирамидных трактов. Градация выраженности нарушения во многом определяется конкретным механизмом развития миелопатии. Так, при компрессионном характере поражении шейного отдела отмечается сочетанность нижнего спастического парапареза и спастико-атрофического пареза рук.
При этом, характерно их изолированное проявление или преобладание расстройств двигательных над чувствительными. Основными жалобами являются: боль в руках с латеральной/медиальной стороны, затруднения при выполнении тонких движений, парестезии в руках, слабость и неловкость в ногах, нарушение походки, развитие нейрогенного мочевого пузыря.
Пирамидный синдром характеризуется несимметричным спастическим тетрапарезом в руках, что обусловлено поражением глубоких пирамидных проводников, отвечающих за верхние конечности. Атрофический синдром проявляется слабостью в мышцах верхних конечностей, атрофиями и фибриллярными подергиваниями, низкими сухожильными рефлексами верхних конечностей.
Цервикальная миелопатия при сосудистом варианте развития миелопатии (сосудистая миелопатия шейного отдела) характеризуется более выраженными и распространенными по длиннику двигательными спинальными сегментарными расстройствами, сочетающихся с ишемией структур входящих в бассейн кровоснабжения передней спинальной артерии (фасцикуляция мышц, отсутствие/снижение рефлексов на руках, амиотрофии).
Наиболее часто встречается дискогенная миелопатия поясничного отдела напрямую обусловленная повреждением межпозвоночного диска, являющейся одним из осложнений остеохондроза позвоночника у пациентов в возрасте после 45 лет и характеризуется хроническим течением. Реже причиной дискогенной миелопатии являются травмы позвоночника и для такой патологии характерно крайне острое течение.
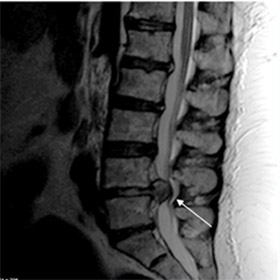
Развитие заболевания обусловлено дегенеративными изменениями в межпозвоночном диске, которые приводят к растяжению/разрыву фиброзного кольца диска и к отрыву его периферических волокон от тел позвонков. Как следствие происходит смещение диска в заднелатеральном направлении, что и приводит к компрессии спинного мозга и прилегающих кровеносных сосудов (фото ниже).
В симптоматике дискогенной поясничной миелопатии наиболее часто встречаются интенсивная радикулярная боль, парезы дистальных отделов ног, снижение мышечной силы ног, нарушение функции органов таза и снижение чувствительности в сакральных сегментах.
Дискогенная миелопатия может осложняется спинальным инсультом (острым расстройством кровообращения) с развитием синдрома трансверзального поражения спинного мозга, для которого характерны сочетание спинальных параличей нижних конечностей с тазовыми нарушениями и глубокой циркулярной гипестезией.
Анализы и диагностика
В основе диагностики миелопатий мануальный осмотр, проверка чувствительности/рефлексов в определенных точках и инструментальные методы исследования, включающие:
- Обзорную/прицельную рентгенографию позвоночного столба в нескольких проекциях.
- Электронейрограмму.
- Компьютерную томографию.
- Магнитно-резонансную томографию.
- Контрастные методы исследования (дискография, пневмомиелография, миелография, веноспондилография, эпидурография).
При необходимости (подозрение на отравление тяжелыми металлами, дефицит витамина В12) назначаются лабораторные исследования. При подозрении на инфицирование проводится спинномозговая пункция.
Лечение
Поскольку миелопатия является обобщенным термином универсальное (стандартизованое для всех случаев) лечение отсутствует, и лечебная тактика определяется в каждом конкретном случае в зависимости от причин, лежащих в основе развития миелопатии. Из общих при принципах лечения можно отметить:
- Для купирования болевого синдрома, уменьшения отека и снижения воспалительного процесса назначаются нестероидные противовоспалительные препараты (Индометацин, Ибупрофен, Ортофен, Диклофенак, Мелоксикам и др.). При сильно выраженной боли, обусловленной сдавлением нервных корешков, назначаются стероидные гормоны (Преднизолон, Дексаметазон и др.).
- Для снятия спазма мышц и уменьшения ощущений назначаются миорелаксанты (Мидокалм, Сирдалуд, Баклосан, Толперизон).
- С целью защиты тканей от гипоксии и нормализации метаболизма используются Актовегин, Церебролизин, Пирацетам и др.
- При наличии инфекции назначаются антибактериальные препараты с учетом чувствительности возбудителя заболевания.
- При необходимости — препараты, восстанавливающие хрящевую ткань (Глюкозамин с хондроитином, Алфлутоп, Артифлекс Хондро, Румалон и др.).
- При ишемической миелопатии назначаются сосудорасширяющие препараты (Папаверин, Кавинтон, Но-Шпа, и нейропротекторы (Глицин, Луцетам, Гамма-аминомасляная кислота, Ноотропил, Гаммалон и др. Для нормализации кровообращения в мелких сосудах и реологических свойств крови — Трентал, Танакан, Пентоксифиллин.
- Для укрепления иммунитета назначаются витаминно-минеральные комплексы или витамины В1 и В6.
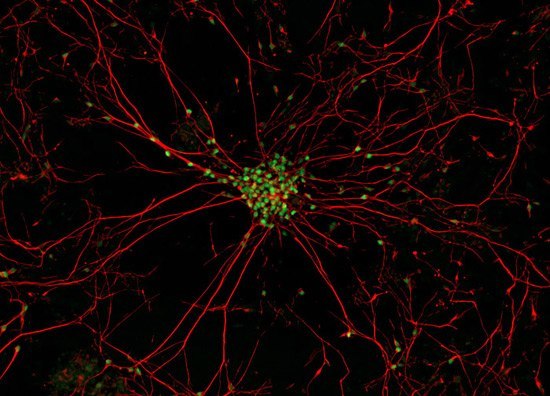
Настоящее фото мотонейрона (двигательного нейрона в передних рогах спинного мозга) - закупорка межсинаптических щелей (зелёные точки) - блокада передачи импульсов через медиаторы (вещества для передачи импульсов) в синапсы (места соединения) отростков нервных клеток.
В центре - тело мотонейрона.
Красные линии -длинные отростки мотонейрона - аксоны и короткие -дендриты.
Причина появления блокады передачи импульсов в межсинаптических щелях учёными мира не найдена. Предположительно - мутация гена,кодирующего фермент передачи этих импульсов через нейромедиаторы - супероксиддисмутазы. (СОД).
Предрасположенность к мутациям гена может иметь наследственный характер по аутосомно-рецессивному типу.
Эндемические (массовые вспышки) случаи этого заболевания зафиксированы у групп военных, живущих в островах на тихом океане. Чаще болеют мужчины от 40 до 60 лет.Следовательно не исключается инфекционная причина развития заболевания.
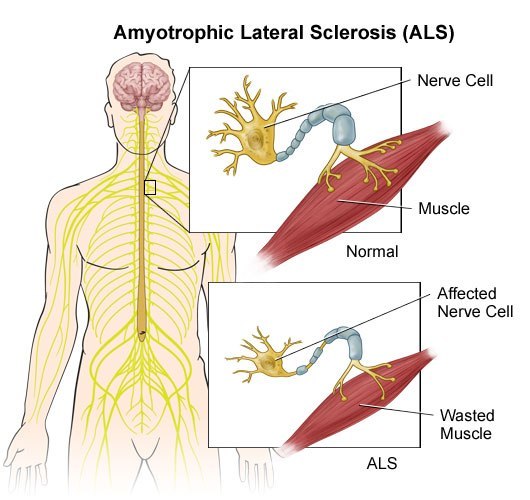
Боковой амиотрофический склероз. (БАС)..
Показано истончение нервных волокон в случае БАС и нарушение иннервации (передачи нервных импульсов) к мышцам. Как следствие - уменьшение работы мышцы и её последующая атрофия. (уменьшение размеров, обратное развитие.)
Блокада передачи нервных импульсов к мышцам (как к поперечно-полосатым которыми мы управляем сами своей волей так и к к гладким, работающим, независимо от нашего сознания, усилий и воли) пищеварительной и дыхательной системы ведёт к смерти из-за невозможности совершать эти жизненно важные моменты работы мускулатуры.
Две статьи из медицинских источников:
1) Теория аксостаза бокового амиотрофического склероза. Аксональная теория бокового амиотрофического склероза
Теория аксостаза основана на анализе патологических процессов, происходящих в аксональном транспорте мотонейронов [Chou S., 1992]. Наибольшими нейронами организма являются двигательные мотонейроны передних рогов спинного мозга и пирамвды Беца. Они должны поддерживать интеграцию дендритов, часто протяженностью более 1 см, и аксон, достигающий 100 см. В аксоне имеются непрерывные потоки, через которые клеточное тело направляет структурные и функциональные белки на периферию и получает обратные сигналы. Ортоградный транспорт бывает 2 видов: а) быстрый — 400 мм в день, идущий в обоих направлениях и транспортирующий связанные с мембраной белки и гликопротеиды, б) медленный — несколько миллиметров в день, транспортирующий сети микрофиламентов, микротрубочек, нейрофиламентов, как компонент "а" (0,1—2 мм в день), а также большой комплекс растворимых белков, как компонент "б" (2—4 мм в день). Ретроградный аксональный транспорт несет эндогенные (аминокислоты, фактор роста нервов) и экзогенные (токсин столбняка, вирус полиомиелита, простого герпеса, бешенства, лектин пероксидазы хрена и др.) субстанции от терминальных аксонов к клеточному телу со скоростью свыше 75 мм в день. Морфологические исследования аксонального транспорта в биоптатах двигательных веточек периферических нервов больных боковым амиотрофическим склерозом выявили уменьшение скорости ретроградного аксонального транспорта и, следовательно, связи терминального аксона с перикарионом [Bieuer A. et al., 1987]. В межреберных нервах больных АБС еще до развития признаков нейрональной дегенерации появляются изменения белков микротрубочек [Binet S. et al., 1988].
Улыраструктурные исследования проксимального аксона и аксонального бугорка мотонейронов переднего рога спинного мозга больных, умерших от бокового амиотрофического склероза [Sasaki S. et al., 1996], показали нарушение быстрого аксонального транспорта. Гладкий эндо-плазматический ретикулум теряет структуру: происходит скопление митохондрий, лизосом, Леви-подобных телец, эозино-фильных и гиалиновых включений, липофусциновых гранул, особенно в аксональном бугорке. Присутствие этих необычных структур является отражением дисфункции аксонального транспорта. Применительно к возможной этиологии АБС еще ранее выдвинута концепция "аксостаза" [Chou S., 1992]. Ней-ротоксические факторы путем ретроградного транспорта избирательно поражают нейрон, создавая феномен "суицидцального транспорта". Ухудшение медленного транспорта в аксоне сопровождается скоплением нейрофиламентов, набуханием проксимального аксона и последующей дистальнои аксональной атрофией, а также вторичной демиелинизацией, характерной для центральной дистальнои аксонопатии или "ретроградного умирания" — "dying back". Определенную значимость в развитии ранних морфологических изменений мотонейронов при АБС имеет теория аутоиммунитета [Smith R. et al., 1996], основанная на появлении антител к зарядам входа кальциевых каналов. Пассивный перенос фракций, содержащих иммуноглобулин, мышам вызывает изменения нервно-мышечных соединений, сходные с таковыми при спорадическом АБС. У животных эти изменения отражают расстройства внутриклеточного Са2+ гомеостаза, и раннее повреждение пластинчатого комплекса в мотонейронах в форме набухания и фрагментации. Иммуноглобулины от больных спорадическим боковым амиотрофическим склерозом вызывают зависимый от Са2+ апоптоз клеток вследствие оксидативных повреждений. Апоптоз, обусловленный иммуноглобулином от указанных больных, регулируется присутствием связанных белков, которые могут модулировать избирательную ранимость нейронов при спорадическом АБС.
2) Боковой амиотрофический склероз
Несмотря на более чем 100-летнее изучение, боковой амиотрофический склероз (БАС) остается фатальным заболеванием центральной нервной системы. Заболевание характеризуется неуклонно прогрессирующим течением с избирательным поражением верхнего и нижнего мотонейронов, что приводит к развитию амиотрофий, параличей и спастичности. До настоящего времени вопросы этиологии и патогенеза остаются невыясненными, в связи с чем не разработаны специфические методы диагностики и лечения этого заболевания. Рядом авторов отмечено повышение частоты встречаемости заболевания среди лиц молодого возраста (до 40 лет).
МКБ-10 G12.2 Болезнь двигательного неврона
ЭПИДЕМИОЛОГИЯ
Боковой амиотрофический склероз дебютирует в возрасте 40 – 60 лет. Средний возраст начала заболевания 56 лет. БАС - болезнь взрослых, и не наблюдается у лиц моложе 16 лет. Несколько чаще заболевают мужчины (отношение мужчины-женщины 1,6-3.0: 1).
БАС является спорадическим заболеванием и встречается с частотой 1,5 – 5 случая на 100 000 населения.
В 90% случаев БАС носит спорадический, а в 10% - семейный или наследственный характер как с аутосомно-доминантным (преимущественно), так и с аутосомно-рецессивным типами наследования. Клинические и патоморфологические характеристики семейного и спорадического БАС практически идентичны.
В настоящее время возраст является основным фактором риска при БАС, что подтверждается нарастанием заболеваемости после 55 лет, и в этой возрастной группе уже не наблюдается различий между мужчинами и женщинами. Несмотря на достоверную связь БАС с возрастом, старение является только одним из предрасполагающих факторов развития патологического процесса. Вариабельность заболевания как в различных возрастных группах, так и среди лиц одного возраста предполагает существование определённых факторов риска: дефицит, или наоборот, наличие определённых нейропротективных факторов, к которым в настоящее время относят: нейростероиды или половые гормоны; нейротрофические факторы; антиоксиданты.
Некоторые исследователи отмечают особо благоприятное течение заболевания у молодых женщин, что подтверждает несомненную роль половых гормонов, в особенности эстрадиола и прогестина, в патогенезе бокового амиотрофического склероза. Подтверждением этому являются: большая частота встречаемости БАС у мужчин до 55 лет (при этом у них отмечается более раннее начало и быстрое прогрессирование заболевания по сравнению с женщинами); с наступлением менопаузы женщины болеют также часто, как и мужчины; единичные случаи заболевания боковым амиотрофическим склерозом во время беременности. К настоящему времени существуют единичные работы по изучению гормонального статуса больных с боковым амиотрофическим склерозом, и ни одной, посвящённой определению концентраций гормонов у молодых пациентов.
Этиология заболевания не ясна. Обсуждается роль вирусов, иммунологических и метаболических нарушений.
В развитии семейной формы БАС показана роль мутации в гене супероксиддисмутазы-1 (Cu/Zn-супероксиддисмутазу, SOD1), 21q22-1 хромосома, выявлен также БАС, связанный с 2q33-q35 хромосомой.
Синдромы, клинически не отличимые от классического БАС, могут возникать в результате:
•опухоли большого затылочного отверстия
•спондилез шейного отдела позвоночника
•артериовенозная аномалия спинного мозга
•бактериальные - столбняк, болезнь Лайма
•вирусные - полиомиелит, опоясывающий лишай
Интоксикации, физические агенты:
•токсины - свинец, алюминий, другие металлы.
Читайте также:
