
Эпизодическая атаксия 2 типа
Эпизодические атаксии, то есть временно возникающая шаткость при ходьбе и хаотичные размашистые движения, относят к генетическим неврологическим заболеваниям. Пациент шатается при ходьбе, может упасть и не встать до окончания приступа. Он не способен взять предмет, застегнуть пуговицу, самостоятельно выпить воды.
Периоды относительного благополучия сменяются приступами раскоординации движений, ребенок не может некоторое время управлять своими движениями. Внешне эпизодические атаксии напоминают проявления эпилептического синдрома, но по сути имеется ряд существенных отличий:
- Во-первых, во время эпизодической атаксии пациент не теряет сознание, а ЭЭГ не регистрирует эпилептической активности.
Клинических вариантов эпилептических пароксизмов крайне много. Только мониторинг ЭЭГ позволит отдифференцировать эпилепсию от другого заболевания и вовремя выставить правильный диагноз, подобрать лечение.
- Во-вторых, атака провоцируется физиологическим или психологическим стрессом: жара или холод, усталость, перенапряжение сил, эмоционально значимые события.
Атаксия способна коснуться и речевого аппарата (отмечаются нарушения речи), тогда мы слышим монотонную, с придыханием, неожиданными паузами и эффектом стаккато, скандированную речь.
Такая разбалансированность произвольных двигательных процессов длится от нескольких минут до нескольких часов, а затем исчезает, в ряде случаев – бесследно до нового пароксизма (приступа). Иногда и между приступами сохраняется небольшая атаксия, потому что ткани мозжечка, центра координации движений, постепенно атрофируются.

Проявляется болезнь в детском или подростковом возрасте, эпизоды атаксии случаются с разной частотой: несколько раз за день, ежедневно, пару раз в неделю, ежемесячно. С возрастом этот показатель урежается.
Эпизодическая атаксия первого типа. Приступ выражен миокимией в сочетании с атаксией. Миокимия или мышечная волна представляет собой периоды сокращений мышцы или ее части, видимыми подергиваниями без совершения движения. Такая атака длится пару минут, впервые проявляясь в раннем детстве. Причиной заболевания является мутация генов, обеспечивающих калий-натриевую передачу:калиевых каналов становится меньше, форма их меняется, клеточные мембраны ощущают дефицит калия, в результате клетки медленно расслабляются после напряжения. Значительно страдает баланс возбуждения и торможения мозжечка, клеток двигательных нейронов спинного мозга и периферических нервов.
Собственно, заболевание развивается у детей среднего возраста, примерно от 7 до 12 лет. Испуг, физические нагрузки, перенесенные соматические заболевания, яркие эмоции, резкие движения – все это способно спровоцировать приступ.
Ребенок испытывает стресс, реагируя на него мышечным напряжением и попыткой действовать быстро. Однако избыточное возбуждение приводит к тому, что клетки часто напрягаются и не успевают расслабиться. В результате развиваются локальные мышечные подергивания (миокимии) и теряется контроль над движениями (атаксия). Вероятность развития эпилепсии у таких детей в десять раз выше, чем обычно.
Клиническая картина
Атаксия сопровождается мозжечковыми нарушениями:
- кружится голова (головокружение);
- двоится в глазах;
- миокимии – изолированные мышечные подергивания;
- тошнит, встречается рвота;
- нарушаются речь и зрение.
Такой приступ длится от нескольких минут до нескольких часов, повторяясь до 10-15 раз за сутки. Помимо атаксических атак в межприступный период отмечаются миокимии – подергивания мышц лица, стоп, голеней, кистей, предплечий.
Эпизоды могут возникать спонтанно и после движения, испуга, на фоне заболевания, сильных положительных и отрицательных эмоций.
Для диагностики необходимо провести генетический анализ, электромиографию, которая обнаружит постоянное напряжение отдельных мышечных волокон. На ЭЭГ при эпизодической атаксии эпи-активности как правило нет, однако во многих случаях заболевание протекает в сочетании с эпилепсией и требует проведения длительной электроэнцефалограммы бодрствования со сном.

Лечение направлено на купирование и предупреждение развития приступов. Оно включает в себя противоэпилептическую и седативную терапию (ацетазоламид, карбамазепин, фенотонин, вальпроат, фенобарбитал). Заболевание неизлечимо, но с возрастом атаки легче и реже.
Мутация гена приводит к изменению белка кальциевого канала (альфа – 1А-субъединица потенциал зависимого кальциевого канала). Этот белок входит в состав клеток мозжечка, формирует просвет каналов, находящихся в этом отделе головного мозга мозжечке, именно они участвуют и в образовании синоптической передачи импульса к нервам и мышцам. Изменения этого же гена встречаются при семейных мигренях, когда к приступам головной боли добавляется небольшая атаксия, эпизодической гемиплегии, спиноцеребеллярной атаксии. В последнем случае атаксия носит постоянный характер, постепенно увеличивая свои проявления. Встречается мутированная часть генов с 80% вероятностью среди членов одной семьи, хотя в целом эпизодическая атаксия – очень редкоезаболевание.
Волнение, физические и психологические стрессы, кофе, алкоголь или психостимулирующие средства, беременность, высокая температура тела или жара провоцируют приступы атаксии, так как на их развитие влияет гормональный фон пациента.
У мальчиков и взрослых мужчин заболевание протекает тяжелее: приступы чаще, нарушения движений, напоминающие судороги, более выражены. Развивается эпизодическая атаксия обычно в детстве, но может впервые появиться и у взрослого человека (дебют болезни от двух до тридцати с небольшим лет).
Клинические проявления:
- разлитая слабость;
- размашистые неконтролируемые движения, подобные судорожному приступу;
- иногда изменение темпа речи;
- головокружение;
- мигрень во время приступа встречается у 50% больных(сильно болит голова);
- тошнота, рвота.
Могут присоединиться следующие симптомы:
Приступ длится дольше, чем при эпизодической атаксии первого типа, от нескольких часов до 1-2 суток. Повторяются пароксизмы с различной частотой: у одних такое состояние развивается несколько раз в неделю, у других – один-два раза в год. Между атаками человек способен вести обычную жизнь, но со временем появляются проявления атаксии: пошатывание, ухудшение координации движений, нарушения баланса тела в сложных позах, а также нистагм и небольшая мышечная слабость.

Девочка пытается ответить, но во рту словно образуется каша, в глазах начинает двоиться, начинает сильно кружиться голова. Разливается невыносимая слабость, и девушка, неловко взмахнув руками, медленно падает на пол.
— Да ты пьяная! – возмущается отец, -Почему у тебя глаза бегают? Вставай немедленно!
Встать она не может – половина тело обмякает. Другой рукой она пытается опереться о ручку двери, но не может ее захватить. Голова болит и кружится, сильно тошнит.
-Что ты кричишь на ребенка? Возмущается мать. – Ты посмотри, у нее же инсульт!
Приезжает Скорая помощь, девочку несут в машину на носилках. В приемном покое голова перестает болеть, движения восстанавливаются, возвращается речь… Дается направление на консультацию к врачу-неврологу по месту жительства.
В диагностический комплекс исследований в обязательном порядке входят: мониторинг ЭЭГ (где фиксируется отсутствие эпи-активности), электромиография (где не отмечается миокимий), генетическое молекулярное исследование, нейрорадиологическое исследование головного мозга или МРТ, свидетельствующие о повреждении мозжечка.
В лечении с успехом применяют ацетазоламид, более известный как диакарб. Он хорошо снимает приступы атаксии, а на фоне постоянного приема их практически не происходит. В то же время диакарб назначается под контролем анализов мочи и крови, не применяется при беременности. К сожалению, этот препарат не влияет на межприступные проявления болезни. Можно добиться длительной ремиссии, но нистагм и шаткость могутоставаться все равно, хотя пациенты мобильны, сами передвигаются и ухаживают за собой.
В таблице №1 представлена сравнительная характеристика эпизодической атаксии первого и второго типов.
Еще реже, чем вышеописанные формы заболевания, встречаются эпизодические атаксии 3 и 4 типов. Эпизодическая атаксия третьего типа начинается в позднем подростковом периоде. Приступ сопровождается головокружением, атаксией и двоением в глазах. Со временем выраженность двигательных нарушений может увеличиваться.
Эпизодическая атаксия четвертого типа объединяет некоторые черты 1 и 2 типа болезнис одной стороны, чувствительна к диакарбу, с другой – сопровождается миокиниями. Начало болезни возможно в любом возрасте в пределах 35 лет жизни, а приступ характеризуется головокружением и непроизвольными движениями. Между атаками встречаются непроизвольные подергивания отдельных мышц, но нистагма и шаткости походки не наблюдается.
Атаксические движения не очень похожи на классический судорожный синдром, но миокимии при эпизодической атаксии первого типа напоминают эпилептический миоклонус. Поэтому для диагностики необходимы периодические ЭЭГ-исследования, тем более что для заболевания первого типа риск присоединения эпилепсии более чем высок.
Источник: глава 15, Эпизодические атаксии. Е.Д.Белоусова, А.Ю.Ермаков. М.Ю.Дорофеева и др.
Эпизодическая атаксия 2-го типа (MIM 108500) была описана, по-видимому, первой из всех форм эпизодических атаксий — в середине прошлого века, и с этого времени заболевание было выявлено в большом числе семей и изолированных случаев в различных популяциях мира (Gancher S.T., Nutt J.G., 1986). Альтернативные названия болезни: эпизодическая атаксия с нистагмом, ацетазолчувствительная эпизодическая атаксия.
Тип наследования — аутосомно-доминантный. Типичный возраст начала симптоматики — от раннего детства до 20 лет. При эпизодической атаксии-2 у больных развиваются мозжечково-вестибу-лярные пароксизмы, сходные с таковыми при эпизодической атак-сии-1: острая атаксия, дизартрия, головокружение, тошнота, рвота, головная боль. Отличия заключаются в следующем:
а) у больных эпизодической атаксией-2 пароксизмы являются более длительными и могут продолжаться от получаса до нескольких часов и даже дней;
б) при эпизодической атаксии-2 в структуру приступа закономерно входит выраженный нистагм, а также (у половины больных) генерализованная мышечная слабость.
Острые эпизоды атаксии при эпизодической атаксии-2 провоцируются эмоциональными и физическими стрессами, лихорадкой и перегреванием организма, приемом алкоголя, кофе, а также пищи, богатой углеводами. Атактические приступы купируются ацетазоламидом (диакарбом) (Baloh R., Winder А.). Без лечения частота приступов может составлять от 3-4 в неделю до 1—2 в год.
В межприступном периоде у больных эпизодической атаксией-2 выявляется целый ряд постоянных неврологических симптомов: нистагм (особенно вертикальный, направленный книзу), нарушение плавных следящих движений глазных яблок, расстройства координации различной степени выраженности. С течением времени выраженность постоянных мозжечковых нарушений может нарастать, и на поздней стадии клиническая картина данной формы эпизодической атаксии начинает все больше напоминать клинику прогрессирующей аутосомно-доминантной спиноцеребеллярной атаксии 6-го типа (СЦА6).
В отдельных случаях у больных эпизодической атаксией-2 описаны также симптомы фокальной дистонии шеи и рук (в том числе во время атактических пароксизмов), эпилептические припадки, а у 10% больных бывают преходящие гемиплегические эпизоды — как при семейной гемиплегической мигрени (Jen J. et al.).
Еще одной особенностью эпизодической атаксии-2 является наличие миастеноподобных проявлений в виде повышенной утомляемости и генерализованной мышечной слабости, что может иметь место у части пациентов в конкретных семьях. Как уже было отмечено, мышечная слабость в 50% случаев может входить в структуру мозжечково-вестибулярных пароксизмов, в части случаев она может непосредственно следовать за пароксизмом и продолжаться 1—3 ч, однако слабость бывает и не связанной непосредственно с эпизодами атаксии. Иногда генерализованная мышечная слабость может на несколько лет предшествовать появлению атактических атак.
Течение болезни чрезвычайно вариабельно. Описано как полное прекращение атактических эпизодов и какой-либо другой симптоматики после 2-го десятилетия жизни, так и медленное неуклонное нарастание мозжечковой атаксии (даже при исчезновении острых пароксизмов), приводящее к перманентной инвалидизации через 15—30 лет от начала болезни.
Сходство поздней стадии эпизодической атаксии-2 с хронической формой СЦА6 усиливается частым обнаружением у больных эпизодической атаксией-2 атрофии червя мозжечка при использовании методов нейровизуализации (КТ, МРТ). Из других методов инструментальной диагностики следует отметить значимость электрофизиологического исследования (ЭМГ), при котором могут выявляться отдельные признаки нарушения нервно-мышечной передачи (Jen J.C, Baloh R.W.).
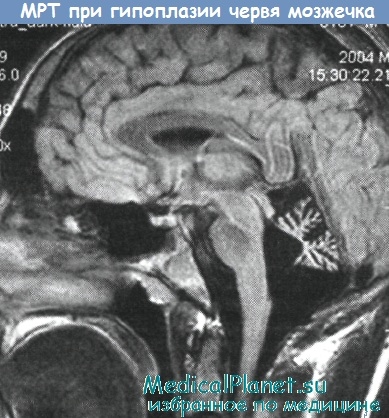
Эпизодическая атаксия-2 обусловлена мутациями гена а1А-субъединицы потенциалзависимого кальциевого канала (CACNL1A4) на хромосоме 19р13.1 (Ophoff R.A. et al.). Причем этот же ген, ответственен за аутосомно-доминантную спиноцеребеллярную атаксию 6-го типа — СЦА6. Дело заключается в том, что при данных аллельных заболеваниях имеют место различные по своему типу мутации: большинство случаев эпизодической атаксии-2 вызываются точковыми мутациями CACNL1A4, ведущими к обрыву трансляции (Ophoff R.A. et al., Denier С. et al.). В то же время, у больных СЦА6 имеет место экспансия тринуклеотидных CAG-повторов в данном гене (Zhuchenko О. et al.).
По мнению O. Zhuchenko с соавт., разные фенотипы атаксии при повреждении одного и того же гена CACNL1А4 обусловлены различным характером нарушения функции потенциалзависимого кальциевого канала у каждой категории больных. Недавно было показано, однако, что эти различия не носят принципиального характера: у некоторых больных с фенотипом эпизодической атаксии-2 заболевание может быть связано с небольшой степенью экспансии CAG-повторов в гене CACNL1A4 (20—23 копии повторов) (Jodice С. et al.).
Интересно отметить, что еще один тип мутаций в этом же гене, CACNL1A4 (точковые миссенс-мутации с заменой аминокислоты), обусловливает развитие совершенно особого и тоже пароксизмального аутосомно-доминантного заболевания — семейной гемиплегической мигрени (Ophoff R.A. et al.).
Полиморфизм комбинаций различных острых пароксизмов и постоянных прогрессирующих неврологических расстройств при данных аллсльных заболеваниях связан, наиболее вероятно, с многообразием функций кальциевых каналов и особенностями их распределения в ЦНС (Battistini S. et al.). Возможность развития при эпизодической атаксии-2 отдельных симптомов миастенического характера свидетельствует также о значимости по-тенциалзависимого кальциевого канала в функционировании нервно-мышечного синапса.
Молекулярная диагностика эпизодической атаксии 2-го типа возможна в случае идентификации мутаций в гене CACNL1A4 у консультируемых лиц (Denier С. et al., Jen J.С. et al.). При этом на первом этапе ДНК-анализа целесообразно исключить экспансию тринуклеотидных CAG-повторов в гене CACNL1A4 в соответствии с протоколом, используемым для прямой ДНК-диагностики СЦА6. В процессе медико-генетического консультирования семьи необходимо принимать во внимание любые пароксизмальные неврологические расстройства у ближайших родственников обследуемого лица (мигрень, эпизоды преходящего гемипареза, приступы мышечной слабости, головокружений и т.п.), поскольку полиморфизм клинических проявлений эпизодической атаксии-2 может носить не только межсемейный, но внутрисемейный характер.
Атаксия — нарушение точности или координации движений.
Атаксия проявляется:
•расстройством походки от легкой степени до невозможности ходить (абазия)
•нарушением равновесия в положении стоя (проба Ромберга) и сидя от легкой степени до невозможности стоять (астазия) и сидеть
•расстройством движений верхних и нижних конечностей в виде промахивания и дрожания при выполнении каких-либо действий - постуральный и интенционный тремор
С анатомо-функциональной и топической точки зрения выделяют мозжечковую, сенситивную, вестибулярную и лобную атаксии.
Поражение:
•полушария мозжечка приводит к развитию атаксии гомолатеральных конечностей
•поражение червя мозжечка — к атаксии туловища
Вестибулярная атаксия возникает при поражении периферического вестибулярного аппарата, кохлеовестибулярного нерва и вестибулярных ядер и путей ствола мозга.
Проявляется:
•головокружением (часто ощущением вращения окружающих предметов – системное головокружение)
•нарушением равновесия при ходьбе, в положении стоя (проба Ромберга) и сидя.
•тошнотой и рвотой (характерно)
•усилением головокружения и неустойчивости при движениях головой.
•отсутствием атаксии в конечностях
•сохранением суставно-мышечное чувства
Лобная атаксия (апраксия ходьбы) наблюдается при поражении лобных долей мозга вследствие повреждения лобно-мосто-мозжечковых связей.
•Наиболее значительна атаксия в контралатеральной очагу поражения ноге, характерны нарушения равновесия, которые могут достигать степени астазии — абазии.
•Обычно отмечается снижение критики, памяти и интеллекта, характерное для поражения лобных долей мозга.
Атаксия может возникнуть при различных неврологических заболеваниях: инсультах, черепно-мозговой травме, рассеянном склерозе, наследственных и идиопатических мозжечковых атаксиях, опухолях и абсцессах мозга, фуникулярном миелозе, полиневропатиях и других болезнях. Атаксия также может развиться при острой и хронической алкогольной интоксикации, отравлениях солями тяжелых металлов и растворителями, гипотиреозе, недостаточности витамина Е, приеме ряда лекарственных средств (карбамазепина, дифенина и др.) и как паранеопластическая реакция при злокачественных новообразованиях (симптоматическая атаксия).
Атаксия (мозжечковая и/или сенситивная) составляет основной клинический синдром ряда дегенеративных заболеваний наследственного, врожденного или идиопатического генеза. В последние годы выявлены генные нарушения и предложена новая классификация наследственных атаксий (на основе обнаруженных генных мутаций): атаксии с аутосомно-рецессивным типом наследования (атаксия Фридрейха, атаксия-телеангио-эктазия — факоматозы, атаксия вследствие недостаточности витамина Е), атаксии с аутосомно-доми-нантным типом наследования (спиноцеребеллярные атаксии 1 —7-го типов, дентато-рубро-паллидарная атрофия, эпизодические атаксии).
Рассмотрим подробнее группу заболеваний с аутосомно-доминантным типом наследования, которые характеризуются повторными острыми эпизодами координаторных нарушений различной длительности, нередко в сочетании с другими, преимущественно мозжечковыми, симптомами.
ЭПИЗОДИЧЕСКИЕ АТАКСИИ
Эпизодические атаксии относятся к группе так называемых острых или пароксизмальных атаксий, так как они протекают приступообразно.
Приступы начинаются в детском или подростковом возрасте и могут отмечаться с частотой от нескольких в день до одного-двух в месяц. В ряде случаев частота приступов снижается по мере взросления.
Большинство пациентов с эпизодическими атаксиями вне атак неврологически здоровы, но у некоторых из них может развиваться и межприступная прогрессирующая атаксия, обусловленная нарастанием атрофических изменений в мозжечке.
С клинической и генетической точки зрения выделяют два основных типа эпизодических атаксий:
•эпизодическая атаксия первого типа
•эпизодическая атаксия второго типа
Эпизодическая атаксия ПЕРВОГО типа (тип 1) (син.:миокимия с периодической атаксией; пароксизмальная атаксия с нейромиотонией; эпизодическая атаксия с миокимией; EA1)
•Этот тип эпизодической атаксии был первым из описанных каналопатий калиевых каналов.
•Тип наследования – аутосомно-доминантный.
•Возможны спорадические случаи.
•Распространенность заболевания неизвестна. Оно считается редким и к настоящему времени описано более ста пациентов.
•Эпизодическая атаксия 1-го типа обусловлена мутациями гена KCNA1, расположенного на 12p13 и кодирующего субъединицу белкового комплекса потенциалзависимого канала калия. Известны по крайней мере 7 различных мутаций.
•При всех мутациях гена KCNA1 патофизиологические механизмы приводят к нарушению функционирования калиевых каналов и к замедлению реполяризации клеточных мембран. Предполагается, что при эпизодической атаксии 1-го типа нарушения обусловлены как уменьшением количества каналов, так и изменением их строения. Дисфункция калиевых каналов приводит к увеличению высвобождения нейротрансмиттеров, в результате чего нарушается баланс возбуждения и торможения в клетках мозжечка.
•Мозжечок страдает более других мозговых структур, так как экспрессия гена максимальна в корзинчатых клетках мозжечка.
•При стрессе и необходимости быстрого движения нарушение равновесия между возбуждением и торможением в мозжечке приводит к ослаблению контроля над движением, и развивается атаксия.
•Миокимия обусловлена изменениями возбудимости мотонейронов передних рогов спинного мозга после сильного мышечного напряжения.
•Миокимия (myokymia; мио- + греч. kyma – волна, волнение; син. псевдофасцикуляция) представляет собой гиперкинез, характеризующийся постоянными или транзиторными сокращениями пучка мышечных волокон, не приводящими к перемещению сегмента конечности.
•Те же патологические изменения (медленная реполяризация и недостаточная гиперполяризация) происходят и в периферических нервах. Поэтому эпизодическая атаксия 1-го типа – заболевание не только центральной, но и периферической нервной системы.
•Кроме того, каналы калия отвечают за возбудимость нейронов и известно, что блокаторы калиевых каналов обладают проконвульсивным эффектом.
•Наблюдающаяся при заболевании задержка реполяризации приводит к тому, что вероятность развития эпилепсии у пациентов с эпизодической атаксией 1-го типа в 10 раз превышает популяционную.
•Заболевание начинается в позднем детстве или в раннем подростковом возрасте.
•В клинической картине у больных отмечаются кратковременные эпизоды мозжечковой атаксии, сопровождающиеся тошнотой, рвотой, головокружением, диплопией, дизартрией, иногда – зрительными нарушениями.
•Эпизоды могут возникать как спонтанно, так и провоцироваться движением, физическими нагрузками, испугом, эмоциями и интеркуррентными заболеваниями.
•Приступы длятся от нескольких секунд до нескольких часов.
•Частота приступов достаточно высокая и иногда может достигать 10–15 в сутки.
•В межприступном и приступном периоде примерно у половины больных могут отмечаться миокимии в лицевой мускулатуре и мышцах дистальных отделов конечностей.
•Степень выраженности миокимии может быть различной – от едва заметных мышечных сокращений до очевидных подергиваний.
•В некоторых семьях наблюдалось сочетание миокимии, атаксических атак и пароксизмального кинезигенного хореоатетоза.
•Кроме молекулярной диагностики единственным информативным обследованием является электромиография, которая регистрирует постоянную активность мотонейронов у всех пациентов.
•При биопсии скелетных мышц, которая была проведена у отдельных пациентов, обнаруживались изменения, характерные для денервации.
•Лечение носит симптоматический характер. Показана частичная эффективность ацетазоламида, карбамазепина, вальпроата, фенитоина и фенобарбитала.
•Ацетазоламид может уменьшить число атак у больных членов одной семьи и оказаться неэффективным у пациентов из другой семьи.
•Точный механизм действия ацетазоламида неизвестен. Предполагается, что он может изменять pН как внутри клетки, так и во внеклеточном пространстве. Возможно, что существуют и другие механизмы, обеспечивающие эффективность препарата.
•Начинают, как правило, со стартовой дозы ацетазоламида, которая равна 125 мг, средняя суточная доза составляет 500–750 мг (в два приема). При необходимости суточная доза может увеличиваться до 1000 мг.
•Эффективность ацетазоламида в целом ниже, чем при эпизодической атаксии 2-го типа.
•Противоэпилептические препараты могут не только снижать частоту атак, но и уменьшать степень выраженности миокимии.
•Если приступы появляются в детстве, то к взрослому возрасту они становятся реже и протекают менее тяжело.
Эпизодическая атаксия ВТОРОГО типа (тип 2) (син.: периодическая вестибулоцеребеллярная атаксия; эпизодическаяатаксия, чувствительная к ацетазоламиду; EA2)
•Распространенность заболевания неизвестна. Оно считается редким.
•Тип наследования – аутосомно-доминантный с 80–90 % пенентрантностью.
•Возможны спорадические случаи.
•Эпизодическая атаксия 2-го типа обусловлена повреждением гена CACNA1A, локализованного на хромосоме 19р и ответственного за синтез белка альфа-1А-субъединицы потенциалзависимого кальциевого канала.
•При обследовании семей с эпизодической атаксией 2-го типа на мутации в гене CACNA1A, в одном случае была выявлена делеция пар нуклеотидов, в другом – сцепленная точечная мутация. Как вариант мутаций de novo определяются нонсенс мутации при спорадических случаях эпизодической атаксии 2-го типа.
•В любом случае эти мутации приводят к усечению или аберрации альфа-1А-субъединицы.
•Альфа-1А-субъединица кальциевого канала является полипептидом, который участвует в формировании полости канала и является компонентом P/Q-типа потенциалзависимых кальциевых каналов, широко представленных в центральной нервной системе.
•Наибольшее количество таких каналов находится в мозжечке, и они являются основными кальциевыми каналами клеток Пуркинье.
•Каналы контролируют возбудимость мембраны и участвуют в высвобождении нейротрансмиттеров.
•Кроме этого, каналы P/Q-типа являются главными синаптическими потенциалзависимыми кальциевыми каналами, ответственными за проведение нервного импульса в нервно-мышечное соединение.
•Определенные типы мутаций CACNA1A обнаруживаются и при других аутосомно-доминантных заболеваниях; 50 % случаев семейной гемиплегической мигрени, включая все случаи с постоянно существующими мозжечковыми симптомами, вызваны миссенс мутациями в гене CACNA1A. Известно, что в 20 % семей с данным заболеванием в межприступном периоде обнаруживаются атаксия и нистагм.
•Патологическая активность кальциевого канала вызывает транзиторную дисфункцию клетки и возникновение эпизодических симптомов.
•Хроническая патология кальциевого гомеостаза может приводить к прогрессирующей дегенерации нервной системы.
•Изучение фенотипических различий между пациентами в одной семье позволяет предположить, что в фенотипическое проявление мутации вносят свой вклад и другие факторы, кроме генетических.
•То, что волнение и стресс вызывают острые атаксические эпизоды, предполагает, что кальциевые каналы или сигнальные функции этих каналов могут регулироваться катехоламинами.
•Гормональный профиль может также играть определенную роль. Известно, что для женщин характерно обострение симптоматики именно во время беременности. У мужчин заболевание, как правило, протекает значительно тяжелее, чем у женщин. Даже в рамках одной семьи для мужчин характерно более раннее начало и более частые и тяжелые приступы, чем у их сестер.
•У пациентов с эпизодической атаксией 2-го типа в клинической симптоматике выделяется жалоба на диффузную слабость во время атаксических приступов. Из-за того что каналы P/Q-типа являются основными пресинаптическими потенциалзависимыми кальциевыми каналами, ответственными за выделение ацетилхолина в нервно-мышечное соединение, мутации гена CACNA1A могут вмешиваться в нормальный процесс проникновения кальция в пресинаптический нервный терминал, приводя к ухудшению воздействия нейротрансмиттеров на нервно-мышечное соединение и тем самым – к мышечной слабости.
•По всей видимости, диффузная мышечная слабость является составной частью клинических проявлений мутаций кальциевого канала.
•Первые симптомы чаще всего появляются в детстве или подростковом возрасте, но описано начало заболевания в очень широких возрастных пределах – от двух до 32 лет.
•Приступы провоцируются стрессом, физической нагрузкой, приемом кофеина и алкоголя. Описана провокация приступов при применении фенитоина, а в одной родословной – провокация атак фебрильными заболеваниями и высокой температурой окружающей среды.
•Характерны приступы атаксии, головокружения, тошноты и рвоты.
•У 50 % пациентов отмечаются приступы мигрени во время атаки.
•Также возможны дизартрия, нистагм, двоение в глазах, дистония и транзиторная гемиплегия.
•У большинства пациентов в период атаки возникает диффузная мышечная слабость.
•Продолжительность атаки – от нескольких часов до нескольких дней.
•Частота атак различна у отдельных пациентов и может колебаться от одного-двух раз в год до трех-четырех раз в неделю.
•Между приступами пациенты чувствуют себя здоровыми, но постепенно развивается и межприступная симптоматика в виде нистагма и координаторных нарушений различной степени выраженности (неустойчивость в позе Ромберга, шаткость при ходьбе и т. д.). Нистагм в межприступном периоде отмечается у 90 % пациентов, атаксия – у 80 %.
•Диагностика эпизодической атаксии 2-го типа опирается в основном на клиническую картину заболевания.
•Решающее значение имеют следующие критерии диагноза:
1.семейная отягощенность по заболеванию с прослеживанием аутосомно-доминантного типа наследования
2.провоцирование атак физической нагрузкой, эмоциональным стрессом, алкоголем и кофеином
3.атаксия и нистагм в момент атаки, часто в сочетании с головокружением, тошнотой и рвотой
4.продолжительность атак, как правило, составляет несколько часов
5.сохранение симптомов атаксии и нистагма в межприступном периоде
6.отсутствие миокимии (клинически и электромиографически).
•При нейрорадиологическом обследовании у пациентов с эпизодической атаксией 2-го типа чаще всего выявляется атрофия мозжечка, преимущественно передней части червя.
•Молекулярная диагностика проводится для подтверждения диагноза при развернутой клинической картине, а также может быть использована для диагноза у асимптомных пациентов.
•Возможна и пренатальная молекулярная диагностика.
•Положительный эффект при применении ацетазоламида (диакарба) был открыт случайно, когда предполагалось, что у пациентов с эпизодической атаксией имеется периодический паралич из-за присущей им выраженной мышечной слабости в период атаки.
•У больных с эпизодической атаксией 2-го типа при назначении ацетазоламида отмечается значительное улучшение вплоть до исчезновения приступов; в связи с чем данная форма иногда называется ацетазолчувствительной наследственной пароксизмальной атаксией
•Атаки могут возобновиться через 48–72 часа после завершения применения препарата.
•Хорошо купируя приступы, ацетазоламид не эффективен в профилактике возникновения межприступных нарушений.
•Применение ацетазоламида противопоказано в первом триместре беременности и ограничено строгими показаниями в остальные триместры.
•Течение заболевания вариабельное. Описаны ремиссии заболевания и через год с момента его начала, и спустя десятилетия. Со временем выраженность межприступных мозжечковых нарушений может нарастать, но пациенты, как правило, всегда передвигаются самостоятельно.
Дифференциальный диагноз эпизодических атаксий необходимо проводить как с наследственными, так и с ненаследственными эпизодическими атаксиями.
К ненаследственным причинам эпизодической атаксии относятся следующие:
•аномалия Арнольда–Киари
•рассеянный склероз
•вертебро-базиллярная недостаточность
•базилярная мигрень
•аномалии лабиринта
Из наследственных заболеваний, способных вызвать эпизодическую атаксию, следует исключить:
•митохондриальные болезни (преимущественно дефицит пируваткарбоксилазы и пируватдегидрогеназы, болезнь Лея)
•нарушения цикла мочевины
•аминоацидурии (болезнь Гартнупа, изовалериановую ацидемию)
Необходимо также отделение эпизодической атаксии 2-го типа от других типов эпизодических атаксий.
Типичные для эпизодических атаксий атаки не очень похожи на эпилептические приступы. Миокимия отчасти может напоминать эпилептический миоклонус, но проведение ЭЭГ разрешает все дифференциально-диагностические проблемы, при том условии, что врач может отделить артефакты, соответствующие мышечным сокращениям (они могут становиться более выраженными после проведения гипервентиляции).
Трудности в диагностике могут представлять те пациенты, у которых есть сочетание эпизодической атаксии и эпилепсии. В данной ситуации невролог просто должен помнить о возможности существования и таких больных.
Читайте также:
