
Классификация нервно мышечных заболеваний по бадаляну
Как известно, к нервно-мышечным заболеваниям относятся первичные поражения мышц, вторичные или неврогенные атрофии, миастения, миотонии и периодический паралич. Наиболее полная, исчерпывающая классификация нервно-мышечных заболеваний была создана в 1967 г. научно-исследовательской группой под руководством J. Н. Walton.
В нее включены все заболевания нервной системы, при которых имеются те или иные нарушения функции мышц:
- заболевания периферического мотонейрона;
- заболевания двигательных нервных корешков;
- заболевания периферических нервов;
- нарушения нервно-мышечной проводимости;
- заболевания мышц;
- некоторые заболевания супраспинальной тонической регуляции, которые могут имитировать нервно-мышечные заболевания.
Таким образом, в основу классификации положен принцип локализации преимущественного дефекта. На ряду с четкими нозологическими формами в классификации нашли место не только соответствующие нервно-мышечные синдромы при тех или иных заболеваниях, но даже симптомы (амиотрофия при поперечном миелите, поражения мышц при газовой гангрене, мышечная слабость при контралатеральных церебральных поражениях).
И. Гаусманова-Петрусевич (1971) разделяет нервно-мышечные заболевания на следующие группы:
- группа миопатий;
- повреждения нервно-мышечного соединения;
- неврогенные амиотрофии.
В группу I автор включает:
- миопатии, характерные в клиническом и генетическом отношении, как прогрессирующие мышечные дистрофии;
- миопатии, характерные в клиническом и генетическом отношении, как миотонические синдромы;
- непрогрессирующие формы миопатий с характерными структурными признаками;
- миопатии, характерные биохимически;
- миопатии, обусловленные гормональными нарушениями;
- воспалительные реакции мышц;
- миопатии, сопутствующие новообразованиям;
- миопатии токсические и медикаментозные.
В группу II включены миастения и миастенические синдромы.
Группу III составляют:
- детская и юношеская спинальная атрофия;
- боковой амиотрофический склероз;
- невральная амиотрофия — болезнь Шарко — Мари — Тута;
- семейная гипертрофическая невропатия Дежерина — Сотта;
- невропатии с характерным нарушением метаболизма.
Нам представляется целесообразным разделить все нервно-мышечные заболевания на две основные группы:
- наследственные,
- ненаследственные, или фенокопии.
Группу I составляют:
- прогрессирующие мышечные дистрофии;
- непрогрессирующие миопатии;
- миотонии;
- периодический паралич;
- неврогенные амиотрофии: невральные и спинальные.
Особое место занимает миастения. В литературе имеются высказывания о возможной наследственной природе или наследственном предрасположении к миастении, хотя наследственный генез заболевания полностью не доказан.
Группа II включает симтоматические формы нервно-мышечных заболеваний, развивающихся на фоне какого-либо основного страдания (системное воспалительное заболевание, эндокринная патология и т. д.).
В литературе приводятся специальные классификации для прогрессирующих мышечных дистрофий. С. Н. Давиденков (1954) предложил следующую классификацию прогрессирующих мышечных атрофий.
Миопатии: юношеская форма, бульбарно-паралитическая; дистальная; раннедетская форма с псевдогипертрофиями; раннедетская форма без псевдогипертрофий.
P. Becker в 1972 г. предложил новую классификацию мышечных дистрофий, разделяющую все миодистрофии по типу наследования. Эта классификация выглядит следующим образом.
- Мышечные дистрофии, сцепленные с полом:
- детский, или злокачественный, тип (Дюшенна);
- доброкачественный, или ювенильный, тип (Беккера — Кинера);
- Х-хромосомная мышечная дистрофия с ранними контрактурами (Эмери — Дрейфуса);
- поздний тип (Хэка — Лаудана);
- летальный тип (Хенсона — Мюллера — Демайера).
- Аутосомно-доминантные дистрофии — плечелопаточно-лицевая форма (Эрба — Ландузи — Дежерина) и др.
- Аутосомно-рецессивные мышечные дистрофии:
- детский тип (псевдодюшенновская);
- ювенильный тип;
- плечепоясной тип;
- взрослый тип.
Широкое распространение получила классификация J. Walton, D. Gardner-Medwin (1974). По этой классификации все миодистрофии разделяются также в основном по характеру наследования мутантного гена.
A. Х-Сцепленная мышечная дистрофия:
- тяжелая (Дюшенна);
- благоприятная (Беккера).
Б. Аутосомно-рецессивная мышечная дистрофия:
- конечностно-поясная, или ювенильная (Эрба);
- детская мышечная дистрофия (псевдодюшенновская);
- врожденные мышечные дистрофии.
B. Лицелопаточно-плечевая (Ландузи — Дежерина).
Г. Дистальная мышечная дистрофия.
Д. Окулярная мышечная дистрофия.
Е. Окулофарингеальная мышечная дистрофия.
Последние несколько форм относятся к аутосомно-доминантным типам наследования с высокой или неполной пенетрантностью?
Л. О. Бадалян (1974) подразделяет прогрессирующие мышечные дистрофии на три основные формы: первичные, вторичные и смешанные.
- Первичные прогрессирующие мышечные дистрофии:
- юношеская (аутосомно-рецессивная) — форма Эрба;
- псевдогипертрофическая (Х-хромосомная) — форма Дюшенна;
- плечелопаточно-лицевая (доминантная) — форма Ландузи — Дежерина;
- редкие и атипичные (офтальмоплегическая, бульбарная, дистальная, миосклеротическая).
- Вторичные прогрессирующие мышечные дистрофии (амиотрофии):
- спинальная амиотрофия Верднига — Гоффманна (аутосомно-рецессивная);
- невральная атрофия Шарко — Мари (доминантная и рецессивная);
- спинальная амиотрофия Арана — Дюшенна;
- редкие и атипичные формы (псевдогипертрофический неврит Дежерина — Сотта, спинальная амиотрофия Кугельберга — Веландер, синдром Русси — Леви).
- Смешанные формы:
- лопаточно-перонеальная форма Давиденкова.
- Миопатические синдромы — фенокопии прогрессирующих мышечных дистрофий:
- при эндокринных заболеваниях;
- при коллагенозах;
- при заболеваниях нервной системы;
- при паразитарных заболеваниях;
- при применении лекарственных препаратов (стероиды, акрихиновые, антибиотики, наркотики);
- при новообразованиях легкого, щитовидной железы, желудка и других органов;
- при некоторых редких заболеваниях (артрогрипоз, болезнь Марфана и т. д.).
Помимо клинических форм, автор предлагает выделять стадии процесса, а именно:
- стадия — с умеренно выраженными двигательными нарушениями (больные могут ходить, выполнять легкую работу, слабость выявляется при нагрузке);
- стадия — с выраженными двигательными затруднениями при ходьбе, подъеме по лестнице, при выполнении физической работы;
- стадия — паралитическая: грубые контрактуры, деформации, самостоятельное передвижение невозможно.
Выделение стадий, по нашему мнению, очень важно для оценки функционального состояния больного и правильного выбора лечения.
Следует заметить, что отнесение нервно-мышечного заболевания к той или иной форме, т. е. уточненная диагностика, довольно часто представляет значительные трудности. Особенно это наблюдается в начальных стадиях процесса, а также в далеко зашедшей фазе с генерализацией мышечных атрофий и развитием грубых вторичных контрактур и ретракций.
Без ЭМГ-исследования невозможно поставить достоверный диагноз при дистальной форме миодистрофии, спинальной псевдомиопатической форме Кугельберга — Веландер и ряде других. В некоторых случаях диагностика возможна только после изучения структуры мышцы с использованием современных гистохимических и электронно-микроскопических методов анализа.
Наследственные нервно-мышечные заболевания(ННМЗ)
Классификация ННМЗ
1. Первичные прогрессирующие мышечные дистрофии (первичные ПМД, миопатии)
2. Вторичные ПМД, или нейрогенные(вторичные) амиотрофии, т.е. спинальные и невральные амиотрофии, обусловленные поражением на различных уровнях (тела или аксона) периферических двигательных нейронов
3. Врожденные непрогрессирующие миопатии
5. Пароксизмальные параличи
Общие признаки ННМЗ:
1.мышечная слабость проявляется симметрично и прогрессирует постепенно
2.мышечная слабость не сопровождается перманентной болью, хотя и возможны болезненные мышечные спазмы – крампи
3.при большинстве форм ПМД слабость раньше проявляется и преобладает в мышцах тазового или плечевого пояса и проксимальных отделах конечностей
4.сухожильные рефлексы снижаются пропорционально выраженности мышечной слабости
5. парестезии, расстройства поверхностной и глубокой чувствительности встречаются нечасто, обычно при невральных амиотрофиях
6. заболевание, как правило, не влияет на функции тазовых органов
ПЕРВИЧНЫЕ ПРОГРЕССИРУЮШИЕ МЫШЕЧНЫЕ ДИСТРОФИИ (МИОПАТИИ)
Относят наследственные болезни, при которых расстройства метаболизма ведут к первичной дистрофии мышц (миопатии)
ХАРАКТЕРНО:
Нарастающая мышечная слабость
Гипотония мышц
Гипотрофия мышц
Сухожильная и периостальная гипорефлексия до арефлексии
Ограничение объема активных движений
Иногда псевдогипертрофия мышц
Отсутствуют фибриллярные и фасцикулярные подергивания
В дебюте чаще страдают мышцы тазового пояса, реже плечевого
Выраженные изменения креатин-креатининового обмена
Снижение механической возбудимости мышц
Миодистрофия псевдогипертрофическая ДЮШЕННА (Х-сцепл. рецессивн. тип)
утрата мышечного белка дистрофина
наиболее злокачественная форма первичных миодистрофий
дебют в раннем детском возрасте
с 2-5 лет уже развивается слабость мышц ТАЗОВОГО пояса, бедер, утиная походка
распространение процесса восходящее (до плечевого пояса)
особенно типична псевдогипертрофия икроножных мышц
со временем возникает слабость мышц лица, языка, глотки, гортани, дыхательных мышц
возможны сухожильные ретракции (чаще пяточного)
развивается кардиопатия
возможен адипозо-генитальный синдром, гипоплазия надпочечников, остеопороз
у 30% отставание в интеллектуальном развитии
высокая степень гиперферментемии (КФК)
Поздняя псевдогипертрофическая Миодистрофия БЕККЕРА-КИНЕРА (Х-сцепл. рецессивн. тип)
дебют от 5 до 20 лет чаще 10-15 лет
течение медленно прогрессирующее
распространение мышечных дистрофий как при миодистрофии Дюшена
поражение сердца выражены меньше
доживают до 30-60 лет, могут иметь детей, интеллект сохранен
повышение активности КФК умеренное
называют мягкой формой миодистрофии Дюшена
качественное изменение белка дистрофина
Миодистрофия ЭМЕРИ-ДРЕЙФУСА-ХОГАНА (Х-сцепл. рецессивн. тип)
дебют с 4-5 летнего возраста
поражение мышц тазового пояса при интактности дистальных отделов конечностей
рано ретракция пяточных сухожилий
псевдогипертрофий нет
позже распространение на плечевой пояс, контрактуры локтевых суставов, др крупн суставов
в поздних стадиях ригидность позвоночника, может быть слабость мышц лица
характерно развитие миокардиодистрофии
интеллект обычно сохранен
иногда доживают до 60 лет
повышение КФК умеренное
ЦЕНТРОНУКЛЕАРНАЯ (миотубулярная) миопатия
форма новорожденных или в период с 5 до 30 лет(поздняя форма)
характерна генерализованная мышечная дистония
в случае поздней формы сначала слабость мышц плечевого и тазового пояса, лица, верхних век, мышц обеспечивающих движения глазных яблок
вытянутый лицевой череп, деформация грудной клетки, Х-образная форма ног
миокардиодистрофия
развитие интеллекта обычное
на ЭМГ изменения первично-мышечного характера
Миодистрофия МЭБРИ (Х-сцепл. рецессивн. тип)
проявляется у мальчиков в пубертатном периоде
слабость мышц тазового пояса и бедер
позже выраженные мышечные псевдогипертрофии
нехарактерны сухожильные контрактуры
кардиомиопатия, липоматоз
интеллект не страдает
течение медленно прогрессирующее
Миодистрофия РОТТАУФА-МОРТЬЕ-БЕЙЕРА (фиброзирующая миопатия) (Х-сцепл. рецессивн. тип)
дебют в детском или юношеском возрасте (чаще 5-12 лет)
выраженные сухожильные ретракции и контрактуры (ограничение тыльн разгиб стоп, затем сгибания шеи, разгибания в локтевых суставах
постепенно формируются патологические позы из-за фиброза мышц
далее невозможно сгибать позвоночник
медленно прогрессируют мышечные гипотрофии
слабость мышц обычно умеренная
преобладают парезы и гипотрофии в лопаточно-плечевой обл. и дистальных отделах ног
псевдогипертрофий нет
характерна кардиомиопатия
интеллект чаще сохранен
выраженная гиперферментемия
нередко доживают до 40-50 лет, умирают от сердечной недостаточности
Ювенильная миодистрофия ЭРБА-РОТА (по аутосомно-рецессивному типу)
дебют в детском или юношеском возрасте чаще в 14-16 лет
конечностно-поясная миодистрофия
прежде всего атрофии мышц тазового пояса
ранний признак утиная походка и др миопатические феномены
в дальнейшем атрофии мышц плечевого пояса, рук (форма ЛЕЙДЕНА-МЕБИУСА)
редко дебют со слабости мышц плечевого пояса (форма ЭРБА)
возможны умеренные псевдогипертрофии, формирование контрактур
при поражении межреберных мышц и диафрагмы – дыхательная недостаточность
мышцы лица чаще не страдают
нередко эндокринопатии
течение вариабельное от мягкого до быстро прогрессирующего
умеренная гиперферментемия
инвалидизация через 10-20 лет
возможна и злокачественная (псевдодюшенновская) форма, дебют в 3-5 лет
Плечелопаточно-лицевая миодистрофия ЛАНДУЗИ-ДЕЖЕРИНА (по аутосомно-рецессивному типу)
дебют чаще к 20 годам, иногда несколько позже
слабость и гипотрофия мышц лица, особенно круговых глаз и рта, мышц плечевого пояса
рано губы тапира, лицо сфинкса, улыбка Джоконды, крыловидные лопатки
далее: слабость передней зубчатой, большой грудной, нижних отделов трапецивидных мышц, широчайшей мышцы спины, двуглавой, трехглавой мышц
далее: слабость перонеальных мышц (появляется степаж)
далее: в меньшей степени проксимальные мышцы нижних конечностей
возможна умеренная псевдогипертрофия икроножных и дельтовидных мышц
сухожильные рефлексы постепенно снижаются
интеллект сохранен
течение относительно мягкое
гиперферментемия умеренная
женщины в 3 раз чаще мужчин болеют
Лопаточно-перонеальная миодистрофия ДАВИДЕНКОВА
1. аутосомно-доминантная форма
проявляется чаще в детстве
иногда во 2-3-м десятилетии жизни
слабость и прогрессирующая гипотрофия мышц плечевого пояса и перонеальной группы
мышц с угасанием сухожильных рефлексов начиная с пяточных, степаж
слабость проксимальных отделов рук и плечевого пояса
возможны дистальные парестезии, гипестезии
как правило не страдают мышцы лица
течение медленно прогрессирующее
возможно развитие мышечных контрактур
повышена активность КФК в крови
2. Х-сцепленная рецессивная форма
дебют в первую декаду жизни иногда с мышечных контрактур
сначала слабость в грудных, дельтовидных мышцах, в мышцах проксимальных отделов рук
позже: перонеальные мышцы
характерно значительное повышение КФК
характерна кардиомиопатия (чаще причина смерти)
Поздняя дистальная миопатия ГОВЕРСА-ВЕЛАНДЕР (миодистрофия ВЕЛАНДЕР) (по аутосомно- доминантный тип с неполн пенетрантностью)
дебют обычно после 20 лет чаще 40-60 лет
медленно прогрессирующее течение
начало со слабости и гипотрофии в мышцах стоп и голеней
позже постепенно мышцы кистей и предплечий
снижаются и исчезают сухожильные и периостальные рефлексы
в поздней стадии поражаются проксимальные мышцы конечностей
чувствительность сохранна
всегда интактны мышцы лица
нет псевдогипертрофий
нехарактерны сухожильные ретракции
возможна кардиомиопатия
Окулярная миодистрофия (наружняя хроническая прогрессирующая офтальмоплегия ГРЕФЕ)
клиника проявляется до 30 лет
медленно нарастающее поражение наружных глазных мышц
протекает, как правило без диплопии и приводит к параличу взора
зрачковые реакции сохранены
первой страдает мышца поднимающая верхнее веко
в далеко зашедшей стадии – двусторонний птоз
со временем могут поражаться мимические, бульбарные, затем скелетные мышцы
Наружная хроническая прогрессирующая офтальмоплегия (миопатия КИЛОХА – НЕВИНА) (по аутосомно- доминантный тип с неполн пенетрантностью)
дебют от 8 мес до 80 лет, чаще на 3-ем десятилетии жизни
медленно нарастающий птоз век, слабость наружных мышц глаза, круговых мышц глаза
(может ассиметрично)
постепенно парез взора вверх, затем в стороны, далее наружная офтальмоплегия
может быть, слабость др мышц лица, жевательных, глотки и гортани (БУЛЬБАРНО-
ОФТАЛЬМОПЛЕГИЧЕСКАЯ форма)
чаще у женщин
Окулофарингеальная миодистрофия (аутосомно-доминантный тип)
прогрессирующая наружная офтальмоплегия + дисфагия и дисфония
1 ВАРИАНТ: характерен птоз век и парез мышц глотки
2 ВАРИАНТ: +парез глазодвигательных мышц, мимических и жевательных мышц,
мышц шеи и проксимальных отделов конечностей
3 ВАРИАНТ: + дистальные отделы конечностей
КФК норма или повышение
ЭМГ признаки миодистрофии
Прогрессирующая офтальмоплегическая миопатия (ядерная с пигментным ретинитом, скротальным языком и снижением интеллекта) проявляется наружным офтальмопарезом/плегией + симптомы по определению
скротальный язык = увеличен с глубокими поперечными бороздами
Окулокраниоскелетный миопатический синдром (синдром КЕРНСА – ШАЯ) (аутосомно-доминантный тип)
дебют до 15 лет в виде наружной офтальмоплегии
далее признаки ПМД на лице, с распространением на мышцы шеи, плечевого и тазового
пояса, проксимальные отделы конечностей
может быть снижение слуха, бульбарный синдром
в ЦСЖ белково-клеточная диссоциация
Поздняя прогрессирующая мышечная дистрофия ШНЕЙДЕРМАНА (аутосомно-доминантный тип)
дебют в 30-40 лет
преимущественно страдают проксимальные отделы конечностей и мышцы лица
прогрессирует медленно
на ЭМГ признаки первичного поражения мышц
Болезнь ВЕРХЕРБЕКА
дебютирует мышечными спазмами в ногах
далее снижение силы в ногах и гипотрофия мышц ног
далее распространение вверх
Мышечная дистрофия с контрактурами ДРЕЙФУСА (по рецессивный, сцепленный с полом тип)
дебют в возрасте 4-5 лет
нарастающая мышечная слабость, преимущественно в мышцах тазового пояса и ног
при ходьбе опирается на большие пальцы стоп
далее формируется поясничный гиперлордоз
особенность: формирование контрактур локтевых и др суставов
псевдогипертрофии нет
нередко поражается миокард, отставание в психическом развитии
Синдром ЛУНДБОРГА (аутосомно-рецессивный тип)
в первые годы жизни олигофрения, катаракта
с пубертатного периода прогрессирующая мышечная слабость преимущественно в прокси-
мальных отделах конечностей
гиперлордоз, утиная походка
половой инфантилизм
со временем слабость мышц шеи, лица, наружных мышц глаза
в поздних стадиях полная обездвиженность
Миопатии митохондриальные
проявляется чаще на 2-м десятилетии жизни
в дебюте: птоз верхних век, наружный офтальмопарез без диплопии (поражение симметр-но)
слабость и похудание сначала в проксимальных мышцах
сухожильная гипорефлексия
длительность прогрессирования процесса вариабельна (мес-десятилетия)
миопатические, невропатические, нервно-мышечные нарушения, вегетативные, обменные и эндокринные расстройства
Нервно-мышечными заболеваниями (НМЗ) является группа патологий, которые передаются на генетическом уровне от родителей детям. Нарушаются мышечные функции, снижается двигательная активность. Появляются характерные клинические симптомы.
Патологические процессы развиваются на фоне нарушений функций нервно-мышечных соединений, при поражении мышц и спинномозговых нейронов, нервов. Правильно подобранная терапия не поможет полностью вылечить человека, но позволит улучшить качество его жизни.
Этиология и неврология
Нервно-мышечные заболевания нарушают нормальную синаптическую передачу импульсов с нервных окончаний к мышечным волокнам. В основе каждого типа патологических изменений лежат аутоиммунные процессы.
Большая группа заболеваний характеризуется не только поражением мышечной ткани, но и периферических нервов, передних рогов спинного мозга. Среди часто диагностируемых патологий выделяют миопатию, миотонию, миастению.
Классификация
Нервно-мышечные заболевания различают по следующим видам:
Описание
Нервно-мышечные заболевания передаются по наследству, чаще появляются у людей, в семье которых были родственники с таким диагнозом.
Приобретенные патологические процессы развиваются в результате гормональных или метаболических нарушений в организме человека. Наблюдается сбой в функционировании иммунной системы. Она вырабатывает клетки, которые атакуют свой организм. Аутоиммунные заболевания приводят к появлению слабости в мышцах.
Нервно-мышечные патологии, сопровождающиеся дистрофическими процессами, поражают следующие области тела человека:
- мышцы;
- нервно-мышечные окончания;
- двигательные нейроны;
- периферические нервы.
При миопатии у человека высоки шансы стать инвалидом в результате утраты подвижности. Все виды нервно-мышечных заболеваний без своевременной терапии влекут за собой последствия. Это может быть не только инвалидность, но и смерть человека.
Стадии и степени
Нервно-мышечные заболевания протекают по стадиям. Определить этап развития патологических процессов поможет врач невролог при помощи медицинской диагностики.
| Название | Описание |
| I стадия | Двигательные нарушения слабо выраженные. |
| II стадия | У больного присутствуют ярко выраженные клинические признаки и наблюдаются серьезные двигательные изменения. |
| III стадия | Пациент не может самостоятельно передвигаться. |
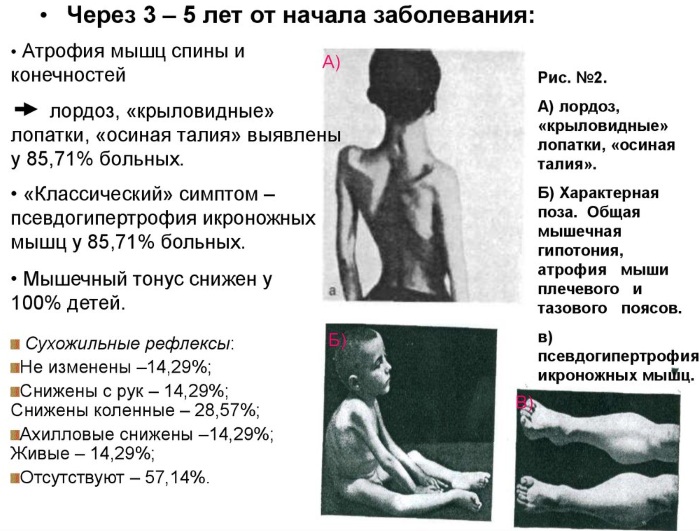
Нервно-мышечные заболевания миопатия Дюшенна на 2 этапе
Клиническая картина зависит от скорости развития патологических процессов и степени тяжести заболевания. Установить точный диагноз поможет врач невролог.
Симптомы
Основной признак нервно-мышечных заболеваний – это слабость мускулатуры. Клиническая картина зависит от области поражения (плечевой пояс, бедра, таз, нижние конечности).
В большинстве случаев у пациентов диагностируют следующие симптомы:
- снижается мышечный объем;
- наблюдаются болезненные спазмы;
- непроизвольно сокращаются мышцы;
- пораженные ткани немеют;
- снижаются сухожильные рефлексы;
- больной ощущает покалывание;
- двоится в глазах (диплопия);
- нарушаются глотательные и дыхательные рефлексы.
При нервно-мышечных заболеваниях опускаются веки, мышечная слабость проявляется симметрично и постепенно прогрессирует. В большинстве случаев при развивающейся мышечной дистрофии слабость возникает в области тазового и плечевого пояса. То же самое касается проксимальных отделов конечностей.
Иногда невральная амиотрофия сопровождается парестезией, нарушением глубокой или поверхностной чувствительности. Клинические признаки нервно-мышечных заболеваний проявляются постепенно. По мере прогрессирования патологических процессов человек теряет способности самостоятельно обслуживать себя. То же самое касается передвижения.
Причины появления
Нервно-мышечные заболевания в большинстве случаев возникают по причине аутоиммунных патологий.
Провоцирующим фактором также являются следующие обстоятельства:
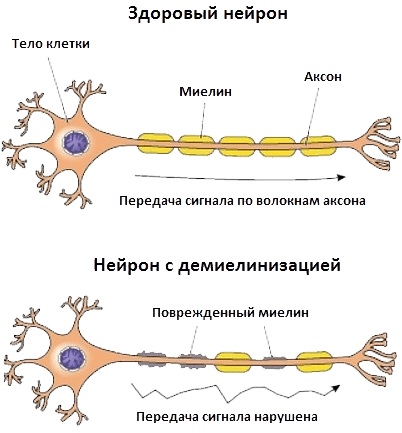
- наследственный фактор;
- поражение периферических нервов и мотонейронов спинного мозга;
- сбои в функционировании нервно-мышечных соединений;
- отравление организма различными веществами;
- врожденный сбой метаболизма;
- патологические изменения в мышцах.
Нервно-мышечные заболевания также развиваются на фоне нарушений работы двигательного нейрона в области ствола головного мозга.
Определить причину и поставить точный диагноз поможет врач невролог. Учитывая состояние пациента, степень развития патологических процессов и индивидуальные особенности человеческого организма, специалист подберет эффективное лечение.
Диагностика
Медицинское обследование позволит врачу установить точный диагноз. Тестирование специалист назначает пациенту, учитывая его жалобы и симптоматику.
Для диагностики нервно-мышечных заболеваний назначаются следующие методы обследования:
Описание

Исследованием состояния сердечной мышцы занимается кардиолог. Специалист назначает не только кардиограмму, но и ультразвуковое исследование (УЗИ) сердца.
Когда необходимо обратиться к врачу
К врачу необходимо обратиться сразу, при появлении первых признаков нарушений в работе мышц. Но если в семье есть родственники с нервно-мышечными заболеваниями, необходимо пройти полное медицинское обследование и понять, насколько высока вероятность появления патологических процессов по наследственной линии.
Диагностикой и лечением занимается врач невролог. Специалист проведет осмотр и подберет максимально информативные методы исследования.
Профилактика
Нервно-мышечные заболевания в большинстве случаев развиваются по причине наследственного фактора. Предупредить патологические изменения невозможно. Женщине во время планирования беременности рекомендуется проходить медицинские обследования, особенно если в семье есть родственники с таким диагнозом.
Диагностические мероприятия также назначаются в период вынашивания малыша на ранних сроках. При высокой вероятности развития нервно-мышечных заболеваний специальная медицинская комиссия советует будущей матери прервать беременность.
Методы лечения
Терапия нервно-мышечных заболеваний осуществляется комплексными методами. Пациентам назначают медицинские препараты, лечебную физкультуру. При отсутствии серьезных противопоказаний, можно использовать рецепты знахарей и целителей. Основная цель терапии – это поддержать мышечные силы и замедлить атрофирующие процессы.
Медикаменты подбирает врач невролог, учитывая результаты медицинской диагностики, степень развития патологических процессов и индивидуальные особенности организма человека.
Самостоятельно не рекомендуется принимать лекарства, поскольку многие препараты вызывают побочные эффекты.
При нервно-мышечных заболеваниях врач назначает следующие медикаменты:
Применение
Лекарства позволяют устранить дефицит энергии и белка, положительно влияют на вещественные обмены в мышечных тканях. Дополнительно назначаются витаминные комплексы.
Нервно-мышечные заболевания можно лечить рецептами знахарей и целителей, но в качестве вспомогательной терапии. Народные средства улучшают качество жизни пациента и общее его состояние. Используемые средства следует обсуждать с лечащим врачом неврологом.
| Название | Рецепт | Применение |
| Овес | Зерна хорошо промыть и залить водой (500 мл). Полученную массу ставят на огонь, доводят до кипения и греют 30 мин. Дальше оставляют на 2 часа, процеживают и принимают по схеме. | Готовое средство рекомендуется принимать внутрь перед едой 4 раза в сутки. Курс терапии продолжается 3 месяца. Затем необходимо сделать перерыв на 30 дней и продолжить терапию. |
| Репчатый лук | Продукт очистить и смешать 200 г с сахаром (200 г), добавить воды (0,5 л). Полученную массу поставить на медленный огонь и греть 1,5 часа. Остудить и добавить 2 ст.л. натурального меда. | Готовое средство рекомендуется принимать по 2 ч.л. 3 раза в сутки. |
| Чеснок | Очистить и измельчить 3 головки чеснока. Добавить 4 лимона, предварительно измельченные. Все компоненты залить медом (1 л) и льняным маслом (200 г). | Полученное средство следует принимать по 1 ч.л. 3 раза в день. |
При нервно-мышечных заболеваниях полезно проводить контрастные ванны для нижних конечностей. После водных процедур ноги рекомендуется укутывать теплым одеялом.
Комплексная терапия нервно-мышечных заболеваний позволяет замедлить их развитие, продлевает период ремиссии и улучшает качество жизни пациента.
Вместе с традиционным и народным лечением больным назначаются следующие методы терапии:

| Название | Описание |
| Физиотерапевтические процедуры | Лечение улучшает проводимость нервных импульсов в мышечных тканях, способствует их питанию. Усиливается кровообращение и вещественный обмен. |
| Массаж | Точечное воздействие помогает повысить тонус мышц. Для достижения лечебного эффекта необходимо провести несколько сеансов на протяжении года. |
| Лечебная физкультура | Гимнастика проводится в специализированном комплексе под наблюдением специалиста. |
Сохранить самостоятельное передвижение пациента позволяют специальные ортопедические приспособления. Лечебная физкультура в виде активных и пассивных движений улучшает состояние больного.
Упражнения следует выполнять регулярно, соблюдая умеренные нагрузки. При нервно-мышечных заболеваниях также рекомендуется плавать. В воде легче выполнять физические упражнения без нагрузки на позвоночник.
Возможные осложнения
Негативные последствия патологических процессов появляются в результате поражения различных внутренних органов и систем организма человека:
Описание
Прогрессирующие патологические процессы также могут спровоцировать искривление позвоночника (кифоз, сколиоз). Больным необходимо носить специальные корсеты. В тяжелых ситуациях или на запущенных стадиях развития нервно-мышечных заболеваний пациенту показано оперативное вмешательство. Решение принимает врач невролог, учитывая состояние человека и индивидуальные особенности его организма.
При нервно-мышечных заболеваниях нарушается двигательная функция, слабеют мышцы. Симптомы постепенно усиливаются на фоне прогрессирующих дистрофических процессов.
Пациенту необходимо пройти полное обследование для постановки диагноза и специально подобранное лечение. Лекарства, средства народной медицины, физиотерапевтические процедуры помогут лишь облегчить жизнь пациенту, но полностью избавить от генетической патологии не смогут.
Оформление статьи: Владимир Великий
Видео о нервно-мышечных заболеваниях
Телесеминар о нервно-мышечных заболеваниях:
Читайте также:
