
Мутация генов атрофия зрительных нервов
а) Аутосомно-доминантная атрофия зрительного нерва и сенсоневральная тугоухость. Описано несколько семей с аутосомно-доминантной атрофией зрительного нерва, сочетающейся с глухотой. Во многих из этих семей не наблюдается других системных или неврологических аномалий. В некоторых, но не во всех этих семьях было выявлено носительство мутаций гена ОРА1, которые в других семьях вызывают изолированную нейрооптикопатию. В одной итальянской семье был идентифицирован новый локус хромосомы 16 (16q21-q22), обозначенный ОРА8, предварительные исследования указывают на то, что в патогенезе также может играть роль митохондриальная дисфункция.
В голландской семье, наследующей доминантную атрофию зрительного нерва и глухоту, мутации ОРА1 были исключены, но была выявлена ранее неизвестная миссенс-мутация гена WFS1 хромосомы 4 (4р16.1), в локусе, мутации которого обычно вызывают аутосомно-рецессивный DIDMOAD-синдром, он же синдром Wolfram, характеризующийся синдромальной нейрооптикопатией, сахарным диабетом, несахарным диабетом и тугоухостью. Аналогично в другой семье, наследующей доминантную атрофию зрительного нерва с тугоухостью и нарушением регуляции глюкозы, при анализе мутаций были исключены мутации генов ОРА1, ОРАЗ, ОРА4 и ОРА5, но была выявлена новая миссенс-мутация гена WFS1 (синдрома Wolfram).
Офтальмоплегия и миопатия развиваются в среднем возрасте. Эта патология представляет собой митохондриальное заболевание, развивающееся вследствие ядерных генетических аномалий.
в) Аутосомно-доминантная атрофия зрительного нерва с рано развивающейся катарактой. В двух семьях из Франции наблюдалось наследуемое по аутосомно-доминантному типу сочетание атрофии зрительного нерва и рано развивающейся катаракты. Мутации гена ОРА1 были исключены, но были выявлены патогенные мутации гена ОРАЗ хромосомы 19 (19q13.2-q13.3), локус этих мутаций обычно вызывает синдром Costeff, синдромальную нейрооптикопатию, наследуемую по аутосомно-рецессивному типу. При скрининговых исследованиях на мутации ОРА3 по поводу моносимптомных доминантных атрофий зрительного нерва в различных семьях не удалось выявить какие-либо патогенные варианты ОРАЗ; вероятно, мутации ОРАЗ, вызывающие доминантную атрофию зрительного нерва, встречаются крайне редко.
г) Аутосомно-рецессивная атрофия зрительного нерва в сочетании с прогрессирующей нейродегенерацией и 3-метилглютакониковой ацидурией III типа (синдром Costeff). При этом аутосомно-рецессивном синдроме, чаще всего встречающемся в еврейских семьях Ирака, тяжелая атрофия зрительного нерва сопутствует экстрапирамидным расстройствам, когнитивным нарушениям, повышению в моче уровня 3-метилглютакониковой кислоты и повышению в плазме уровней 3-метилглютаровой кислоты. Вызывающий заболевание ген был локализован на хромосоме 19 (19q13.2—q13.3) и получил название ОРА3.

DIDMOAD. Диск зрительного нерва атрофичен,
видны ретинальные кровоизлияния, вызванные сахарным диабетом.
д) Аутосомно-рецессивная атрофия зрительного нерва в сочетании с ювенильным сахарным диабетом, несахарным диабетом и тугоухостью (синдром Wolfram). Этот синдром включает в себя ювенильный сахарный диабет и прогрессирующее ухудшение зрения на фоне атрофии зрительного нерва, почти всегда сопровождающихся несахарным диабетом, нейросенсорной тугоухостью или и тем, и другим (вследствие чего был предложен эпоним DIDMOAD — diabetes insipidus, diabetes mellitus, optic atrophy, deafness, т. e. несахарный диабет, сахарный диабет, атрофия зрительного нерва, глухота).
Сахарный диабет развивается в течение первого или второго десятилетия жизни и обычно предшествует развитию атрофии зрительного нерва. Однако у нескольких пациентов атрофия зрительного нерва и ухудшение зрения являлись первыми проявлениями синдрома. На ранних стадиях заболевания острота зрения может оставаться нормальной, несмотря на легкую дисхроматопсию и атрофию зрительного нерва. На поздних стадиях развивается тяжелое ухудшение зрения. При периметрии выявляются генерализованное сужение и центральные скотомы поля зрения. Всегда развивается тяжелая атрофия зрительного нерва, может наблюдаться маленькая или средних размеров экскавация ДЗН. И ухудшение слуха, и несахарный диабет дебютируют в первом или во втором десятилетии жизни и могут протекать в очень тяжелой форме. У половины пациентов присутствует атония мочевыводящих путей, сопровождающаяся рецидивирующими инфекциями мочевых путей, нейрогенным недержанием и даже смертельными осложнениями.
Другие системные и неврологические нарушения включают в себя атаксию, осевую ригидность, судороги, стартл-миоклонус, тремор, нарушение моторики желудочно-кишечного тракта, нарушение вестибулярных функций, центральное апноэ, нейрогенный коллапс верхних дыхательных путей, птоз, катаракту, пигментную ретинопатию, ирит, снижение слезопродукции, зрачки Adie, офтальмоплегию, недостаточность конвергенции, паралич вертикального взора, нистагм, умственную отсталость, психиатрические нарушения, маленький рост, первичную гонадную атрофию, другие эндокринные аномалии, аносмию, мегалобластную и сидеробластную анемию, аномалии при электроретинографии и повышение содержания белка в цереброспинальной жидкости. При лучевых исследованиях и на вскрытии у некоторых пациентов выявляются обширные атрофические изменения и мальформации развития коры, что указывает на диффузное нейродегенеративное расстройство с преимущественным поражением среднего мозга и моста. Если синдром сопровождается анемией, терапия тиамином может облегчить анемию и снизить потребность в инсулине.
В нескольких семьях методом анализа сцепления удалось локализовать ген синдрома Wolfram на хромосоме 4 (4р16.1), который был обозначен как WFS1; были описаны точечные мутации и делеции этого гена. Продукт данного гена — вольфрамин — белок эндоплазматического ретикулума, участвующий в регуляции уровня внутриклеточного кальция. Идентифицирован второй вызывающий синдром Wolfram ген, локализованный на другом плече хромосомы 4 (4q22-24), он получил обозначение CISD2. У больных наблюдается повышенная кровоточивость и пептические язвы. Нокаут гена CISD2 у мышей вызывает развитие Wolfram-подобного синдрома, сопровождающегося преждевременным старением вследствие поражения митохондрий. В общем, многие из сопутствующих аномалий, описанных при синдроме Wolfram, обычно встречаются у пациентов с предположительно митохондриальной патологией, особенно у больных с синдромами хронической прогрессирующей наружной офтальмоплегии.
На основании этих данных было высказано предположение, что фенотип Wolfram, возможно, представляет собой не специфическую аномалию, а является проявлением различных патогенных ядерных или митохондриальных генетических дефектов, в конечном итоге запускающих общий механизм митохондриальной дисфункции. К тому же большинство случаев синдрома Wolfram классифицировались как спорадические или наследуемые по рецессивному типу, последний вывод делался на основании экспрессии синдрома у сиблингов (что, как сейчас известно, также соответствует и материнскому типу наследования).
е) Спастическая параплегия, атрофия зрительного нерва и нейропатия (синдром SPOAN — spastic paraplegia, optic atrophy, neuropathy). Аутосомно-рецессивное нейродегенеративное заболевание было описано клинически как сочетание непрогрессирующей врожденной атрофии зрительного нерва, дебютирующей в младенческом возрасте спастической параплегии, дебютирующего в детстве прогрессирующего поражения двигательных и чувствительных аксонов, дебютирующей в третьем десятилетии жизни дизартрией, выраженных слуховых стартл-реакций, прогрессирующих контрактур суставов и деформации позвоночника. Выявлено сцепление с хромосомой 11q13, но патогенный ген все еще не установлен.
ж) Врожденная мозжечковая атаксия, умственная отсталость, атрофия зрительного нерва и аномалии кожи (congenital cerebellar ataxia, mental retardation, optic atrophy, skin abnormalities— CAMOS). Непрогрессирующая аутосомно-рецессивная врожденная атаксия в сочетании с атрофией зрительного нерва, тяжелой умственной отсталостью и аномалиями строения кожи; заболевание сцеплено с локусом хромосомы 15 (15q24-q26), но патогенный ген все еще не выявлен.
з) Глухота, дистония и нейрооптикопатия (deafness, dystonia, optic neuropathy—DDON, синдром Mohr-Tranebjaerg). Это Х-сцепленное заболевание манифестирует в старшем детском возрасте сенсоневральной тугоухостью, дистонией и атаксией, после чего в возрасте около двадцати лет наблюдается атрофия зрительного нерва, а до пятидесяти лет развиваются снижение интеллекта и психиатрические нарушения. Прогноз для зрения неблагоприятный, большинство пациентов слепнут в возрасте около сорока лет. Причиной заболевания являются мутации гена TIMM8АХ-хромосомы (Xq22), продукт гена локализуется в межмембранном пространстве митохондрий. Было выявлено нарушение биохимических процессов в митохондриях.
и) Осложненная наследственная инфантильная атрофия зрительного нерва (синдром Behr). Описание синдрома Behr включает в себя манифестирующую в детстве атрофию зрительного нерва, в сочетании с различными нарушениями пирамидного тракта, атаксией, умственной отсталостью, недержанием мочи и полой стопой. Синдром обычно наследуется по аутосомно-рецессивному типу, поражаются представители обоего пола. Ухудшение зрения, от умеренного до тяжелого, обычно развивается в возрасте младше десяти лет, часто сопровождается нистагмом. В большинстве случаев по прошествии периода детства патология не прогрессирует. При лучевых исследованиях выявляются диффузные симметричные изменения белого вещества. У некоторых пациентов с синдромом Behr клиническая картина может напоминать проявления наследственной атаксии. Вероятно, синдром Behr является гетерогенным заболеванием, развивающимся вследствие различных этиологических, в том числе генетических, факторов.
к) Прогрессирующая энцефалопатия с отеком, гипсаритмия и атрофия зрительного нерва (синдром РЕНО — progressive encephalopathy with edema, hypsarrhythmia, optic atrophy). Описаны прогрессирующая энцефалопатия, манифестирующая в первые полгода жизни, вслед за которой развиваются тяжелая гипотония, судороги с гипсаритмией, тяжелая умственная отсталость, гиперрефлексия, транзиторный или персистирующий отек лица и тела, атрофия зрительного нерва. Последняя обычно выявляется на первом или втором году жизни, часто наблюдается нистагм. Дефект метаболизма еще не установлен; вероятно, заболевание наследуется по аутосо-мно-рецессивному типу. Данную патологию можно считать формой синдрома Behr, который, вероятно, представляет собой гетерогенную группу заболеваний.
л) Нейрооптикопатия как проявление наследственных дегенеративных заболеваний или патологии развития. Нейрооптикопатия может сопутствовать различным наследственным дегенеративным заболеваниям или системным нарушениям развития. В таблице ниже перечислены основные проявления наиболее часто встречающихся из них.
Наследственные нейрооптикопатии — группа заболеваний, при которых дисфункции зрительного нерва имеют наследственные причины, выявленные на основании семейного наследования или генетического анализа. Клиническая вариабельность одного заболевания как внутри-, так и межсемейная, часто затрудняет распознавание и классификацию этой патологии.
Наследственные нейрооптикопатии часто классифицируются по типу наследования; чаще всего это аутосомно-доминантный, аутосомно-рецессивный и материнский (наследование через митохондриальную ДНК (мтДНК, mtDNA)). Наследуемые по одному типу нейрооптикопатии не обязательно вызываются одним генетическим дефектом. Аналогично, различные генетические дефекты могут являться причиной идентичных или схожих фенотипов — одни из которых передаются по общему типу наследования, другие — нет. И наоборот, один генетический дефект может вызывать развитие различных клинических проявлений, хотя тип наследования должен совпадать.
Также зачастую причиной единичного случая является предполагаемый или подтвержденный генетический дефект, что делает невозможным классификацию на основании типа наследования.
Наследственные нейрооптикопатии обычно манифестируют симметричным двусторонним безболезненным снижением центрального зрения. Многие заболеваний этой группы характеризуются поражением папилломакулярного нервного пучка, что вызывает развитие центральных или центроцекальных скотом. Точная локализация первичного дефекта структур ганглиозных клеток и их аксонов и патофизиологические механизмы поражения зрительного нерва остаются неизвестными, но в большинстве, если не во всех, случаях наследственных нейрооптикопатий центральную роль в патогенезе играет митохондриальная дисфункция. Поражение зрительного нерва обычно персистирует и может прогрессировать. На момент развитии атрофии уже имеется обширное поражение зрительного нерва.
При некоторых наследственных нейрооптикопатиях наблюдается изолированная дисфункция зрительного нерва. При других поражение зрительного нерва всегда сопровождается различными неврологическими и системными аномалиями, некоторые наследственные заболевания, например мультисистемные дегенерации, характеризуются первичными неврологическими или системными нарушениями и могут сопровождаться и атрофией зрительного нерва. В настоящей главе наследственные нейрооптикопатии разделены на три основных группы:
1. Заболевания, преимущественно не сопровождающиеся неврологическими или системными проявлениями.
2. Заболевания, часто сопутствующие неврологическим или системным проявлениям.
3. Состояния, при которых нейрооптикопатия обычно диагностируется как вторичное поражение на фоне общего заболевания.
По мере открытия других специфических генетических дефектов, вероятно, будут изменяться и наши представления о фенотипах и классификации заболеваний этой группы.
Хотя большинство пациентов затрудняются точно определить возраст начала ухудшения зрения, большинство больных отмечают появление жалоб со стороны зрения в возрасте между четырьмя и десятью годами жизни, к одиннадцатилетнему возрасту отмечают ухудшение зрения 58-84% пациентов. Изредка при развитии тяжелого поражения нарушение зрительных функций и сенсорный нистагм наблюдаются уже в дошкольном возрасте. Многие пациенты не подозревают об имеющихся у них нарушениях зрения, пока у них при обследовании членов семьи больного не диагностируется атрофия зрительного нерва.
Такие случаи обычно свидетельствуют о незамеченном дебюте заболевания в детстве, легкой степени зрительной дисфункции, отсутствии ночной слепоты и медленном прогрессировании. Снижение остроты зрения обычно одинаково невелико на обоих глазах. Острота зрения больных варьирует от 20/20 до 20/800, всего лишь около 15% пациентов имеют остроту зрения 20/200 или ниже. Снижение остроты зрения до движения руки или световосприятия встречается крайне редко. Отмечается выраженная межсемейная и внутрисемейная вариабельность тяжести нарушения зрительных функций.
Хотя и не так быстро развивающиеся и не настолько тяжелые, как при наследственной нейрооптикопатии Leber, нарушения зрения при доминантной атрофии зрительного нерва примерно в 50% случаев не позволяют больным управлять механическим транспортным средством. Классическим для больных доминантной атрофией зрительного нерва является нарушение восприятия синего — желтого цветов, но чаще всего наблюдается смешанное нарушение цветовосприятия. При периметрии пациентов с доминантной атрофией зрительного нерва характерно наличие центральных, парацентральных или центроцекальных скотом. Двустороннее снижение чувствительности может симулировать сдавление хиазмы. Атрофия зрительного нерва у пациентов с наследуемой по доминантному типу нейрооптикопатией может протекать незаметно, проявляться лишь побледнением височной половины ДЗН или же вызывать изменения всего диска. Наиболее характерным симптомом является выраженная треугольная экскавация височной половины диска зрительного нерва; иногда таким больным ставится ошибочный диагноз глаукомы.
Незаметное прогрессирующее ухудшение зрительных функций происходит приблизительно у 50-75% пациентов.
Считается, что доминантная атрофия зрительного нерва представляет собой первичную дегенерацию ганглиозных клеток сетчатки. Причиной большинства случаев доминантной атрофии зрительного нерва (50-60%) являются мутации гена ОРА1, локализующегося в длинном плече хромосомы 3 (3q28-q29). Этот ядерный ген широко экспрессируется в митохондриях сетчатки и кодирует динамин-связанный белок, фиксированный на внутренней мембране крист митохондрий. Идентифицировано более двухсот патогенных мутаций гена ОРА1, в том числе миссенс, нонсенс, делеции/инсерции и мутации сплайсинга. Однако моносимптомный фенотип доминантной атрофии зрительного нерва является генетически гетерогенным состоянием, его причиной могут быть дефекты и других локусов. В одной немецкой семье, наследующей доминантную атрофию зрительного нерва, патогенный локус ОРА4 был картирован на хромосоме 18 (18q12.2-12.3), а в двух не родственных семьях из Франции локус ОРА5 был картирован на хромосоме 22 (22q12.1-13.1).
Но ни продукты этих генов, ни их функции описаны не были. Интересно, что при анализе сцепления была выявлена связь глаукомы нормального давления с полиморфизмом гена OPAL.
Классическая картина доминантной атрофии зрительного нерва включает в себя только атрофию зрительного нерва и не сопровождается другими неврологическими или системными нарушениями. Однако давно было известно, что в некоторых семьях у некоторых родственников развивалась классическая картина доминантной атрофии зрительного нерва, а у других больных членов семьи наблюдались другие клинические проявления, такие как сенсоневральная тугоухость и даже хроническая прогрессирующая наружная офтальмоплегия, атаксия, миопатия, периферическая нейропатия и, редко, фенотип, соответствующий наследственной спастической параплегии.
б) Аутосомно-рецессивная атрофия зрительного нерва. Эта форма атрофии зрительного нерва манифестирует при рождении или развивается в раннем возрасте, диагноз обычно ставится в возрасте младше 3-4 лет. Считается, что заболевание наследуется по аутосомно-рецессивному типу, родители пациентов часто являются близкими родственниками. Снижение остроты зрения тяжелое, может развиваться слепота с сенсорным нистагмом. При периметрии определяется сужение полей зрения различной степени, часто выявляются парацентральные скотомы.
Диски зрительных нервов полностью атрофичны, часто формируется тотальная экскавация. По результатам одной офтальмоскопии трудно отдифференцировать это заболевание от инфантильной дегенерации сетчатки, поэтому большое значение для диагностики приобретает электроретинография.
В одной семье французского происхождения, где имелись близкородственные браки, изолированная рецессивная атрофия зрительного нерва была связана с дефектом хромосомы 8 (8q21-q22), патогенный ген был обозначен как ОРА6, хотя продукт гена и его функции еще предстоит определить. В нескольких семьях из Северной Африки с близкородственными браками, наследующих аутосомно-рецессивную атрофию зрительного нерва, изолированную и сопровождающуюся легкой (часто бессимптомной) сенсоневральной тугоухостью, был выявлен дефект гена хромосомы 11, обозначенного ОРА7 (11q14.1—q21), кодирующего трансмембранный белок митохондрий.
в) Х-сцепленная атрофия зрительного нерва. Семейства с подтвержденным сцепленным с полом наследованием атрофии зрительного нерва встречаются крайне редко, особенно редко — наследующие первичную моносимптомную атрофию зрительного нерва. В двух семьях была выявлена сцепленность с одним и тем же локусом Х-хромосомы (Xp11.4-Хр11.2),патогенный ген получил название ОРА2, хотя продукт гена и его функцию еще предстоит установить. В других семьях с предположительно Х-сцепленным типом наследования атрофия зрительного нерва чаще сопровождается другой неврологической и системной патологией.
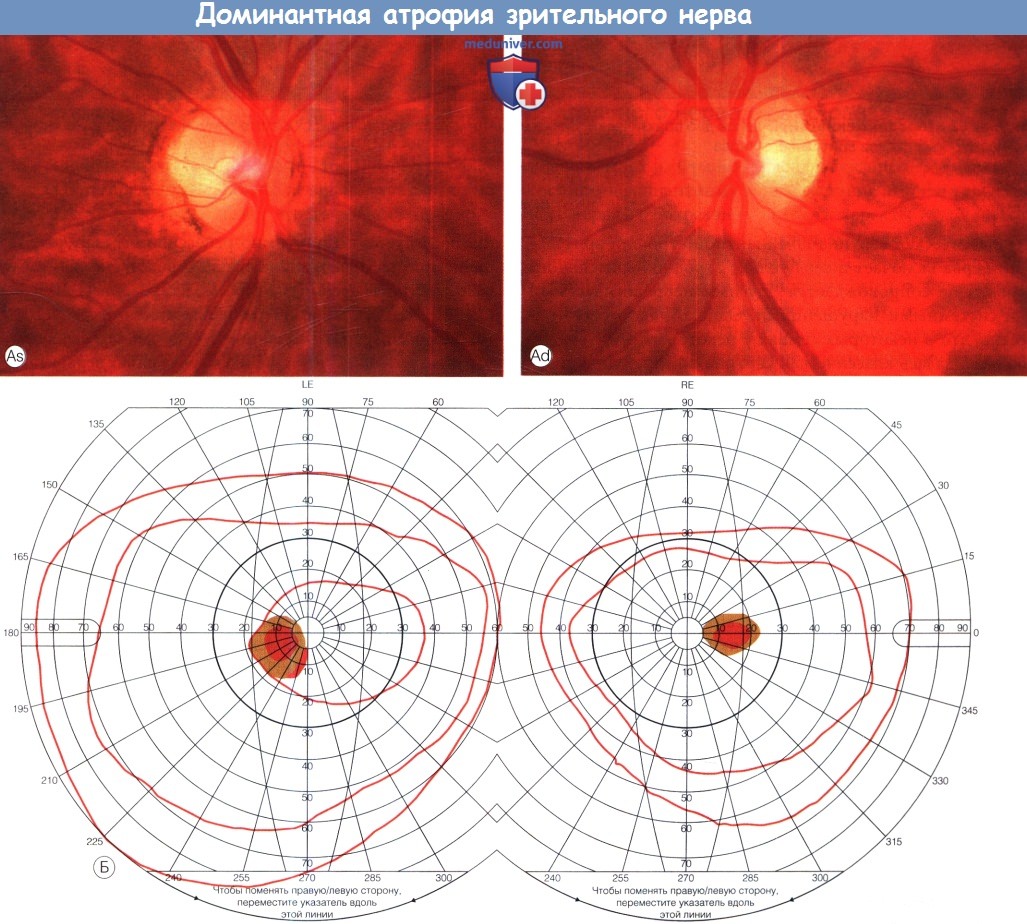
Доминантная атрофия зрительного нерва.
(А) При офтальмоскопии на обоих глазах наблюдаются побледнение височной половины диска зрительного нерва (ДЗН), обширная экскавация.
(Б) При периметрии по Goldmann определяются двусторонние центроцекальные скотомы (снижение чувствительности). 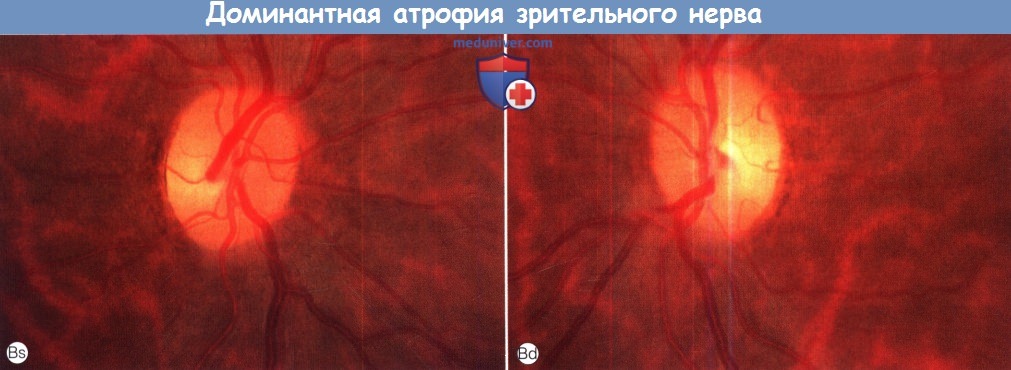
(В) Глазное дно практически здоровой матери того же пациента. Обратите внимание на побледнение височной части обоих дисков зрительного нерва.
*Импакт фактор за 2018 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Читайте в новом номере
В статье приводится описание клинического случая атрофии зрительного нерва вследствие патогенной гомозиготной мутации в 3-м экзоне гена C19orf12 (chr19:30193863AACAGCCCCCCG> A, rs515726204), частота которой в контрольной выборке ExAC составляет 0,0074%. При этой мутации возникает сдвиг рамки считывания в 69-й позиции (p.Gly69fs, NM_001031726.3), которая обычно приводит к нейродегенерации с накоплением железа в мозге (NBIA), тип 4 (OMIM: 614298). В представленном в данной статье клиническом наблюдении ведущей жалобой пациентов являлось ухудшение зрения, при магнитно-резонансной томографии выявлено только расширение области турецкого седла. У 7-летнего мальчика за 2 года значительно снизилось зрение без видимой причины, очковая коррекция не давала улучшения зрения до 100%. При обследовании была выявлена частичная атрофия зрительных нервов, проведены консультации неврологом, нейроофтальмологом. Выявлены гиперрефлексия, изменение походки, незначительная задержка речевого развития. Другой характерной клинической неврологической симптоматики не наблюдалось. В статье приводится описание подробной офтальмологической клинической картины, обсуждается диагностическая и лечебная тактика.
Ключевые слова: частичная атрофия зрительного нерва, ЧАЗН, нейродегенерация с накоплением железа в мозге, NBIA, мутация, ген, C19orf12.
Для цитирования: Иванова М.Е., Кадышев В.В., Атарщиков Д.С. и др. Вариант фенотипа частичной атрофии зрительного нерва вследствие мутации в гене С19orf12 (нейродегенерация с накоплением железа в мозге (nbia)). РМЖ. Клиническая офтальмология. 2020;1:32-36. DOI: 10.32364/2311-7729-2020-20-1-32-36.
Case of phenotype of optic nerve atrophy due to mutation in С19orf12 gene (neurodegeneration with the brain iron accumulation (nbia))
M.E. Ivanova 1 , V.V. Kadyshev 2 , D.S. Atarshchikov 3 , I.V. Zolnikova 4 , N.P. Akchurina 4 , N.K. Serova 5 , F.A. Konovalov 6 , E.R. Lozier 6 , E.A. Pomerantseva 7 , N.V. Vetrova 7 , D. Barh 8 , L.M. Balashova 9 ,
J.M. Salmasi 10
1 LLC “Oftalmic”, Moscow, Russian Federation
2 Research Centre for Medical Genetics, Moscow, Russian Federation
3 Central Clinical Hospital under Presidential Affairs, Moscow, Russian Federation
4 Moscow Helmholtz Research Institute of Eye Diseases, Moscow, Russian Federation
5 N.N. Burdenko National Medical Research Center of Neurosurgery, Moscow, Russian
Federation
6 Independent Clinical Bioinformatics Laboratory, Moscow, Russian Federation
7 Center for Genetics and Reproductive Medicine “Genetiko”, Moscow, Russian
Federation
8 Institute of Integrative Omics and Applied Biotechnology (IIOAB), Bangalore, India
9 Non-profit partnership International Scientific and Practical Center for the Proliferation
of Tissues of Russia, Moscow, Russian Federation
10 Pirogov Russian National Research Medical University, Moscow, Russian Federation
The article describes the clinical case of optic atrophy due to a homozygous mutation in exon 3 of the C19orf12 gene (chr19: 30193863AACAGCCCCCCG> A, rs515726204), the frequency of which in the ExAC control sample is 0.0074. With this mutation, a frameshift occurs at 69-th position (p.Gly69fs, NM_001031726.3), which usually leads to neurodegeneration with the brain iron accumulation (NBIA), type 4 (OMIM: 614298). In described clinical case the main complaint of patient was visual impairment, with magnetic resonance imaging patient revealed only the expansion of the sellar fossa. The vision of 7-year-old boy decreased significantly for 2 years without any apparent reasons, spectacle correction did not give an improvement in vision to 100%. During the examination partial atrophy of the optic nerves was revealed, consultations were conducted with a neurologist, neurophthalmologist. Hyperreflexia, gait changes, and a slight delay in speech development were also revealed. No other clinical neurological symptoms were observed. The article describes a detailed ophthalmic clinical picture, discusses diagnostic and therapeutic tactics.
Keywords: optic nerve atrophy, neurodegeneration with the brain iron accumulation, NBIA, mutation, gene, C19orf12.
For citation: Ivanova M.E., Kadyshev V.V., Atarshchikov D.S. et al. Case of phenotype of optic nerve atrophy due to mutation in С19orf12 gene (neurodegeneration with the brain iron accumulation (nbia)). Russian Journal of Clinical Ophthalmology. 2020;20(1):–36. DOI: 10.32364/2311-7729-2020-20-1-33-36.
Введение
Мутации в гене C19orf12 характеризуются первоначально изменениями походки, сопровождающимися прогрессирующим спастическим парезом, прогрессирующей дистонией (которая может быть ограничена руками и ногами или быть более общего характера), психоневрологическими нарушениями (например, эмоциональной лабильностью, депрессией, беспокойством, импульсивностью, галлюцинациями, невнимательностью и гиперактивностью), снижением когнитивных способностей [1, 2]. Дополнительные ранние признаки могут включать дисфагию, дизартрию, атрофию зрительного нерва, аксональную нейропатию, паркинсонизм и недержание кала/мочи. Обычно пациенты доживают до взрослого возраста, но не оставляют потомства. Конечная стадия заболевания характеризуется тяжелой деменцией, спастичностью, дистонией и паркинсонизмом [3, 4]. Мутации в гене C19orf12 характерны для населения восточноевропейских стран [5, 6].
Клиническое наблюдение
У мальчика 7 лет в течение 2 лет значительно снизилось зрение без видимой причины, очковая коррекция не давала улучшения зрения до 100%. При обследовании была выявлена частичная атрофия зрительных нервов, проведены консультации неврологом, нейроофтальмологом. Выявлена гиперрефлексия, изменение походки (ходит на носочках), незначительная задержка речевого развития. Другой характерной клинической неврологической симптоматики не наблюдалось. Телосложение нормостеничное, рост 140 см, вес 30 кг, роды в срок, стигм дизэмбриогенеза не выявлено, перинатальные инфекции отрицают.
При офтальмологическом обследовании (2018 г.)
Vis OD = 0,5 sph -0,50 = 0,8
Vis OS = 0,5 sph -0,50 = 0,8
Авторефрактометрия:
OD sph -0,50 cyl 0,00
OS sph -0,25 cyl -0,5 ax 5
Пневмотонометрия OD 22 мм рт. ст., OS 18 мм рт. ст.
Status oculorum: положение глаз в орбите правильное, глазные структуры сформированы полностью, врожденных пороков органа зрения не наблюдается, движение глаз в полном объеме, девиация по Гиршбергу 0 градусов, передняя камера средней глубины, влага чистая, радужная оболочка в норме, зрачки в центре округлой формы, диаметр OD=OS=4 мм, прямая и содружественная фотореакция зрачков в норме, хрусталики в задней камере, прозрачны, в стекловидном теле единичные плавающие помутнения.
Офтальмоскопия: глазное дно: OU — диски зрительного нерва бледные, монотонные, границы четкие, сосудистый пучок в центре, сосуды сужены, макулярный и фовеальный рефлексы четкие, периферия сетчатки без патологии (тропикамид 1,0%) [7].
По данным электроретинографии (ЭРГ) выявлено нарушение электрогенеза наружных и средних слоев в макулярной зоне (рис. 1).
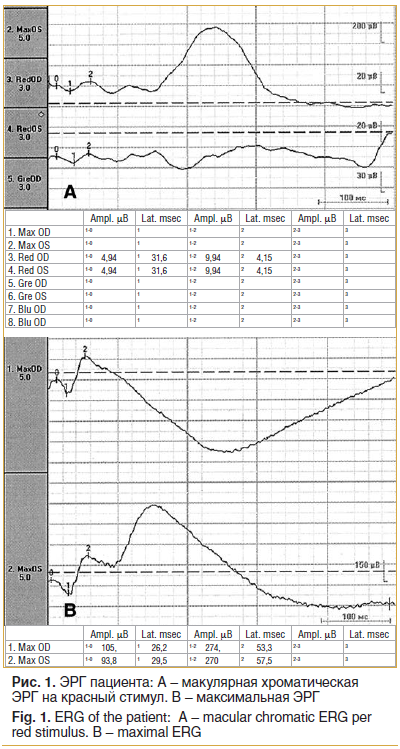
Зрительные вызванные потенциалы: на паттерн, амплитуда снижена на 30 угловых минут OD, на 60 угловых минут OS.
Оптическая когерентная томография (ОКТ) показала равномерное истончение наружных слоев сетчатки. Макулярный рефлекс слабо дифференцируется (рис. 2).

Периметрия (результаты недостоверны): сужение полей зрения до 20–25 градусов.
Цветовосприятие: сохранено.

При неврологическом обследовании выявлена гиперрефлексия, изменение походки (ходит на носочках), незначительная задержка речевого развития. Другой характерной клинической неврологической симптоматики не наблюдается.
Консультация нейроофтальмолога: патология сетчатки, зрительного нерва врожденного характера.
Консультация генетика: родители здоровы, национальность — русские, родственный брак полностью исключить не представляется возможным. У пробанда три старших брата (рис. 4), ни у кого в семье схожих заболеваний не было. У самого старшего брата выявлена миопия - 4,0 дптр, а также склонность к нервным тикам. Других заболеваний глаз, нервной системы и других органов у старших братьев пробанда не обнаружено.

Результаты генетического анализа. По результатам проведения полноэкзомного секвенирования (Illumina Novaseq 6000 с применением набора Agilent SureSelect Human AII Exon V.7, биоинформатическая обработка проприетарными алгоритмами Независимой лабораторией клинической биоинформатики, Москва) обнаружена описанная ранее мутация в гомозиготном состоянии в 3-м экзоне C19orf12 (chr19:30193863AACAGCCCCCCG>A, rs515726204), приводящая к сдвигу рамки считывания, начиная с 69-го кодона (p.Gly69fs, NM_001031726.3). Мутация описана в гомозиготной форме и компаунд-гетерозиготной форме вместе с другими мутациями у пациентов с нейродегенерацией с накоплением железа в головном мозге, тип 4 (OMIM: 614298) [3, 5]. Частота мутации в контрольной выборке ExAC составляет 0,0074%. По совокупности сведений мутацию следует расценивать как патогенную.
Обсуждение
Белок C19orf12 локализован в основном в мембране эндоплазматического ретикулума (ЭПР) и в мембране митохондрий. Вторичная и третичная структура C19orf12 представлена на рисунке 5. Предсказание трансмембранных областей выполнено с помощью MEMSAT, трансмембранные области прогнозируются в спиральных структурах PSI-Pred, выделены зеленым цветом (рис. 5А). Трехмерная (3D) модель полноразмерного белка C19orf12 гомологичного N-домену MgtE представлена в виде лент (рис. 5В). Белок показан оттенком цвета от синего до красного, от N- до С-конца соответственно. Точки и стрелки обозначают области, которые, как ожидается, соединяют домен с мембраной [8].

На рисунке 5С показан предиктивный анализ укороченной вследствие мутации у пробанда версии белка C19orf12 с применением инструмента Phyre2 для построения модели. Функциональный анализ in silico предсказывает значительное нарушение функции белка вследствие имеющейся мутации, что объясняет наблюдаемую клиническую картину, однако нельзя исключить резкое прогрессирование заболевания в этом случае.
ТМ — трансмембранный домен митохондриальной мембраны и мембраны эндоплазматического ретикулума, где наблюдается расположение большей части белка C19orf12. Пространственное строение нормального белка C19orf12 указано на рисунке 5В. Предиктивный in silico анализ возможного изменения структуры белка у данного пациента вследствие frameshift-мутации в 3-м экзон chr19:30193863AACAGCCCCCCG>A, rs515726204 со сдвигом рамки считывания, начиная с 69-го кодона p.Gly69fs с применением online программного инструмента Phyre2 [9], показан на рисунке 5С.
Прогноз: средней тяжести.
Медико-генетическое консультирование: риск рождения в семье ребенка с NBIA (Neurodegeneration With The Brain Iron Accumulation — нейродегенерация с накоплением железа в головном мозге) составляет 25%. Вопрос деторождения в данной семье не актуален.
Лечение и наблюдение: в данном случае целесообразно применять хелатные препараты, например деферипрон (не зарегистрирован в РФ) для уменьшения накопления железа в нейронах головного мозга с целью снижения потенциального прогрессирования неврологической симптоматики. Назначено регулярное применение хелатных препаратов железа в течение 2 лет с дальнейшей оценкой клинического и функционального состояния, проведение МРТ в динамике.
Заключение
Приведенный клинический случай иллюстрирует важность своевременной и точной диагностики пациентов с различными формами аномалий сетчатки и зрительного нерва, в т. ч. с применением технологий секвенирования нового поколения. В данном случае по результатам генетического анализа был значительно уточнен клинический и генетический диагноз нейродегенерации с накоплением железа в мозге (NBIA), что дает шанс замедлить прогрессирование заболевания с помощью препаратов, влияющих на метаболизм железа в организме. Несмотря на то, что некоторые клиницисты [10] низко оценивают перспективы патогенетически направленной терапии хелатами, применение препаратов, влияющих на различные звенья патогенеза данного заболевания, в свете подробных знаний о нем позволит облегчить течение болезни. Также с учетом свойств гена C19orf12 вероятность применения генной терапии в будущем с хорошим терапевтическим эффектом довольно высока.
Сведения об авторах:
1 Иванова Марианна Евгеньевна — к.м.н., врач-офтальмолог, руководитель, ORCID iD 0000-0002-1089-4293;
2 Кадышев Виталий Викторович — к.м.н., старший научный сотрудник, врач-генетик, офтальмолог, ORCID iD 0000-0001-7765-3307;
3 Атарщиков Дмитрий Сергеевич — к.м.н., врач-офтальмолог, ORCID iD 0000-0003-4401-9099;
4 Зольникова Инна Владимировна — д.м.н., врач-офтальмолог, старший научный сотрудник, ORCID iD 0000-0001-7264-396X;
4 Акчурина Наталья Павловна — к.м.н., врач-офтальмолог, ORCID iD 0000-0001-9155-791X;
5 Серова Наталья Константиновна — д.м.н., нейроофтальмолог, главный научный сотрудник, ORCID iD 0000-0003-0148-7298;
6 Коновалов Федор Андреевич — к.б.н., биоинформатик, руководитель, ORCID iD 0000-0001-6414-436X;
6 Лозиер Екатерина Ричардовна — биоинформатик, ORCID iD 0000-0003-2901-0539;
7 Померанцева Екатерина Алексеевна — к.б.н., врач-генетик, ORCID iD 0000-0002-6765-7133;
7 Ветрова Наталья Владимировна — к.м.н., врач-генетик, ORCID iD 0000-0001-5142-6851;
8 Бар Дебмала — PhD, руководитель отдела анализа омиксных данных, ORCID iD 0000-0002-2557-7768;
9 Балашова Лариса Маратовна — д.м.н., врач-офтальмолог, ORCID iD 0000-0001-9349-7092;
10 Салмаси Жеан Мустафаевич — д.м.н., профессор, заведующий кафедрой патофизиологии, ORCID iD 0000-0001-8524-0019.
6 Лаборатория клинической биоинформатики. 123181, Россия, г. Москва, ул. Маршала Катукова, д. 21, корп. 1.
8 Центр геномики и прикладной генной технологии. 560032, Индия, г. Бангалор, ул. Чоланаяканахалли, д. 209.
10 ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России. 117997, Россия, г. Москва, ул. Островитянова, д. 1.
Контактная информация: Иванова Марианна Евгеньевна, e-mail: info@oftalmic.ru.
About the authors:
1 Marianna E. Ivanova — MD, PhD, Head of CRO, ORCID iD 0000-0002-1089-4293;
2 Vitaly V. Kadyshev — MD, PhD, Senior Researcher ophthalmologist, geneticist, ORCID iD 0000-0001-7765-3307;
3 Dmitry S. Atarshchikov — MD, PhD, ophthalmologist, ORCID iD 0000-0003-4401-9099;
4 Inna V. Zolnikova — MD, PhD, ophthalmologist, Senior Researcher, ORCID iD 0000-0001-7264-396X;
4 Natalya P. Akchurina — MD, PhD, ophthalmologist, ORCID iD 0000-0002-6726-4612;
5 Natalya K. Serova — MD, PhD, neuroophthalmologist, Chief Researcher, ORCID iD 0000-0003-0148-7298;
6 Fedor A. Konovalov — PhD, Head of bioinformatics department, ORCID iD 0000-0001-6414-436X;
6 Ekaterina R. Lozier — bioinformatician, ORCID iD 0000-0003-2901-0539;
7 Ekaterina A. Pomerantseva — MD, PhD, geneticist, ORCID iD 0000-0002-6765-7133;
7 Natalya V. Vetrova — MD, PhD, geneticist, ORCID iD 0000-0002-7012-2359;
8 Debmalya Barh — PhD, Head of OMICs analysis department, ORCID iD 0000-0002-2557-7768;
9 Larisa M. Balashova — MD, PhD, Professor, Head of the Center, ORCID iD 0000-0001-9349-7092;
10 Jean M. Salmasi — MD, PhD, Professor, Head of Patho-physiology Department, ORCID iD 0000-0001-8524-0019.
1 LLC “Oftalmic”. 47/3–3, Leningradsky Prospekt, Moscow, 125167, Russian Federation.
2 Research Centre for Medical Genetics. 1, Moskvorechie str., Moscow, 115478, Russian Federation.
3 Central Clinical Hospital for Presidential Affairs. 15, Marshala Timoshenko str., Moscow,121359, Russian Federation.
4 Moscow Helmholtz Research Institute of Eye Diseases. 14/19, Sadovaya-Chernogryazskaya str., Moscow, 105062, Russian Federation.
5 N.N. Burdenko National Medical Research Center of Neurosurgery. 16, 4-th Tverskaya-Yamskaya str., Moscow, 125047, Russian Federation.
6 Independent Clinical Bioinformatics Laboratory, 21/1, Marshala Katukova str., Moscow, 123181, Russian Federation.
7 Center for Genetics and Reproductive Medicine “Genetiko”. 3/1, Gubkina str., Moscow, 119333, Russian Federation.
8 Institute of Integrative Omics and Applied Biotechnology (IIOAB). 209, Cholanayakanahalli str., Bangalore, 560032, India.
9 Non-profit partnership International Scientific and Practical Center for the Proliferation of Tissues of Russia. 29/14, Prechistenka str., Moscow, 119034, Russian Federation.
10 Pirogov Russian National Research Medical University. 1, Ostrovityanov str., Moscow, 117513, Russian Federation.
Contact information: Marianna E. Ivanova, e-mail: info@ oftalmic.ru. Financial Disclosure: Marianna E. Ivanova is the collaborator of LLC “Oftalmic”. Fedor A. Konovalov, Ekaterina R. Lozier are employees of the Independent Clinical Bioinformatics Laboratory, Ekaterina A. Pomerantseva, Natalya V. Vetrova are employees of the “Genetiko” Center. Other authors declare that there is no conflict of interests. Received 14.09.2019.
Только для зарегистрированных пользователей
Читайте также:
