
Нервно мышечные заболевания геномед
Панель объединяет в себе заболевания, характеризующиеся не только нарушением функции скелетных мышц, но и патологией нейронов и проводящих путей. Также в панель включена группа нейродегенеративных и метаболических заболеваний, обладающих схожими клиническими проявлениями, что позволяет облегчить дифференциальный поиск.
Распространенность нервно-мышечных заболеваний колеблется в диапазоне 2.4-33.8 на каждые 100 000 человек, а заболеваемость варьирует по разным данным от 1.16 до 19 на каждый миллион человек в год.
Ключевыми симптомами, характерными для заболеваний из этой группы, являются мышечная слабость, атрофия, дистония, миотония, формирование контрактур, фасцикуляции.
Панель включает 391 ген, ответственный за возникновение нервно-мышечных заболеваний: изолированные и синдромальные моторно-сенсорные и сенсорно-вегетативные нейропатии, врожденные структурные миопатии и мышечные дистрофии, метаболические миопатии, спинальные мышечные атрофии, миастении, боковой амиотрофический склероз, миотонии и периодический паралич, а также ряд генов, ассоциированных с заболеваниями со схожими клиническими проявлениями.
Исследуемые гены: AARS, ABCC9, ABHD12, ABHD5, ACAD9, ACADM, ACADS, ACADVL, ACTA1, ACTC1, ACTN2, ACVR1, ADCY6, AGL, AGRN, AIFM1, AKAP9, ALDOA, ALG13, ALG14, ALG2, ALS2, AMPD1, ANG, ANK2, ANO5, ARHGEF10, ASAH1, ATL1, ATL3, ATP2A1, ATP2A2, ATP7A, ATXN2, B3GALNT2, B4GAT1, BAG3, BICD2, BIN1, BMPR2, BSCL2, C10orf2, C12orf65, C9orf72, CACNA1C, CACNA1D, CACNA1S, CACNB2, CALR3, CAPN3, CASQ1, CASQ2, CAV3, CCDC78, CCT5, CFL2, CHAT, CHCHD10, CHKB, CHMP2B, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, CHST14, CLCN1, CNTN1, CNTNAP1, COL12A1, COL4A1, COL6A1, COL6A2, COL6A3, COLQ, COX6A1, CPT2, CRYAB, CSRP3, CTDP1, DAG1, DAO, DARS, DCAF8, DCTN1, DES, DHTKD1, DMD, DNA2, DNAJB2, DNAJB6, DNAJC3, DNM2, DNMT1, DOK7, DPAGT1, DPM2, DPM3, DPP6, DSC2, DSG2, DSP, DST, DTNA, DYNC1H1, DYSF, ECEL1, EGR2, EMD, ENO3, ERBB3, ERBB4, ERCC3, ERCC4, ERCC5, ERCC6, ERCC8, ETFA, ETFB, ETFDH, EXOSC3, EXOSC8, EYA4, FAM134B, FBLN5, FBN1, FBN2, FBXO38, FGD4, FHL1, FIG4, FKBP10, FKBP14, FKRP, FKTN, FLNC, FUS, FXN, G6PC, GAA, GAN, GARS, GBE1, GDAP1, GFPT1, GJA1, GJA5, GJB1, GLA, GLE1, GMPPB, GNB4, GNE, GPD1L, GRN, GYG1, GYS1, GYS2, HADH, HADHA, HADHB, HARS, HCN4, HEXA, HINT1, HK1, HNRNPA1, HNRNPA2B1, HNRNPDL, HOXD10, HSPB1, HSPB3, HSPB8, IGHMBP2, IKBKAP, INF2, ISCU, ISPD, ITGA7, JPH1, JPH2, JUP, KARS, KBTBD13, KCNA5, KCND3, KCNE1, KCNE2, KCNE3, KCNH2, KCNJ2, KCNJ5, KCNQ1, KIF1A, KIF1B, KIF5A, KLHL40, KLHL41, LAMA2, LAMA4, LAMB2, LAMP2, LARGE, LDB3, LDHA, LITAF, LMNA, LMOD3, LPIN1, LRP4, LRSAM1, MAMLD1, MARS, MATR3, MED25, MEGF10, MFN2, MPZ, MSTN, MTM1, MTMR14, MTMR2, MUSK, MYBPC1, MYBPC3, MYF6, MYH14, MYH2, MYH3, MYH6, MYH7, MYH8, MYL2, MYL3, MYLK2, MYOT, MYOZ2, MYPN, NALCN, NDRG1, NEB, NEFH, NEFL, NEXN, NGF, NPPA, NTRK1, OPA1, OPA3, OPTN, ORAI1, PABPN1, PDHA1, PDK3, PFKM, PFN1, PGAM2, PGK1, PGM1, PHKA1, PHKA2, PHKB, PHKG2, PIEZO2, PIP5K1C, PKP2, PLEC, PLEKHG5, PLN, PLOD2, PMP22, PNPLA2, POLG, POLG2, POMGNT1, POMGNT2, POMK, POMT1, POMT2, PRDM12, PRKAG2, PRPH2, PRPS1, PRX, PSEN1, PSEN2, PTRF, PUS1, PYGL, PYGM, RAB7A, RAPSN, RAX2, RBCK1, RBM20, REEP1, RNF170, RRM2B, RYR1, RYR2, SBF1, SBF2, SCN10A, SCN11A, SCN1B, SCN3B, SCN4A, SCN4B, SCN5A, SCN9A, SCO2, SEPN1, SEPT9, SETX, SGCA, SGCB, SGCD, SGCE, SGCG, SH3TC2, SIGMAR1, SIL1, SLC12A6, SLC16A1, SLC22A5, SLC25A20, SLC37A4, SLC5A7, SMCHD1, SMN1, SMN2, SNAP25, SNTA1, SOD1, SOX10, SPEG, SPG11, SPTLC1, SPTLC2, SQSTM1, STAC3, STIM1, SUCLA2, SURF1, SYNE1, SYNE2, SYT2, TARDBP, TAZ, TBK1, TCAP, TDP1, TFG, TGFB3, TIA1, TK2, TMEM43, TMEM5, TMPO, TNNC1, TNNI2, TNNI3, TNNT1, TNNT2, TNNT3, TNPO3, TPM1, TPM2, TPM3, TRAPPC11, TRDN, TRIM2, TRIM32, TRPA1, TRPM4, TRPV4, TTN, TTR, TUBA4A, TYMP, UBA1, UBQLN2, VAPB, VCL, VCP, VMA21, VRK1, WNK1, YARS, YARS2, ZBTB42, ZC4H2
Материал для исследования: Венозная кровь с ЭДТА 2 мл.
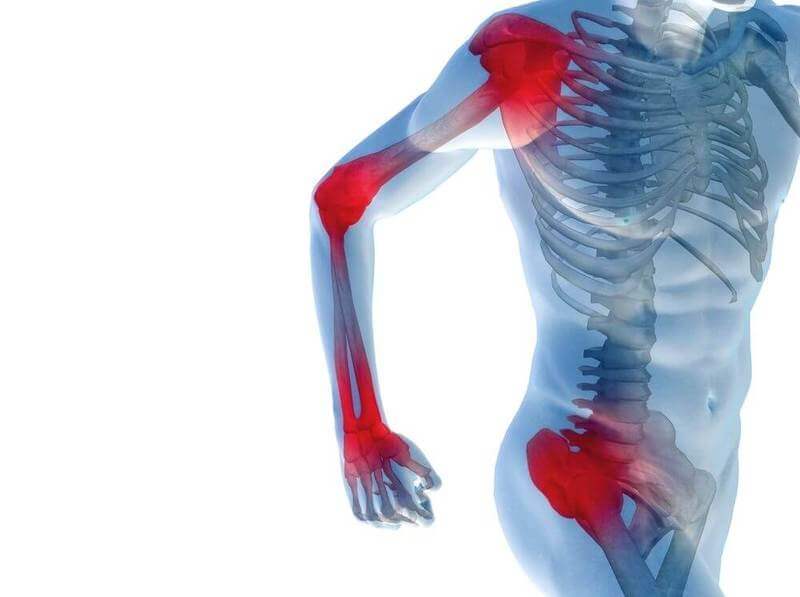
Миопатии, или болезни мышц, — группа заболеваний, объединяющих различные нарушения в строении и метаболизме мышечной ткани и сопровождающихся снижением силы и ограничением двигательной активности. Они проявляются мышечной слабостью, развитием мышечной атрофии, снижением сухожильных рефлексов и мышечного тонуса.
Виды миопатий
Болезни мышц относятся к группе нервно-мышечных заболеваний. Они делятся на первичные и вторичные.
Первичные миопатии обусловлены генетически детерминированными нарушениями в функционировании митохондрий и ионных каналов миофибрилл, а также сбоями в продуцировании белков или ферментов, которые регулируют метаболизм мышечной ткани. Наследование дефектного гена, обуславливающего болезни мышц, может происходить по доминантному, рецессивному и сцепленному с Х-хромосомой типу. Зачастую внешние факторы выступают в роли пусковых механизмов развития болезни.
Приобретенные болезни мышц могут возникать на фоне эндокринных патологий, хронических интоксикаций, авитаминоза, мальабсорбции, тяжелых хронических заболеваний. В эту группу входят аутоиммунные миастении, посттравматические и метаболические миопатии, краш-синдром и т. д.
Наследственные болезни мышц
Большинство наследственных нервно-мышечных заболеваний наследуются аутосомно-рецессивно и характеризуются высоким риском передачи по наследству. Такие болезни подразделяют:
- на первично-мышечные заболевания (прогрессирующие мышечные дистрофии, врожденные структурные миопатии, лице-плече-лопаточная дистрофия, дистальные мышечные дистрофии и т. д.);
- метаболические миопатии (связанные с дефицитом фосфорилазы, кислой мальтазы, фосфофруктокиназы, карнитина и т. д.);
- болезни мотонейрона (спинальная мышечная атрофия, боковой амиотрофический склероз);
- миотонии и периодический паралич;
- заболевания периферических нервов;
- болезни нервно-мышечных синапсов и др.
Диагностика болезней мышц
Для установки диагноза используются электрофизиологические методы обследования: электромиография и электронейрография. Они помогают дифференцировать болезни мышц от миелита, миелопатий, опухолей спинного мозга, нарушений спинномозгового кровообращения.
Биохимический анализ крови при миопатиях показывает повышенный уровень альдолазы, АЛТ, КФК, АСТ, ЛДГ. Биохимический анализ мочи может обнаружить высокий уровень креатинина.
В целях диагностики болезней мышц проводят их биопсию. Морфологическое исследование образцов может обнаружить беспорядочно разбросанные атрофированные миофибриллы среди сохранных и гипертрофированных мышечных волокон, участки замещения мышечной ткани на жировую или соединительную.
МРТ мышц позволяет визуализировать волокна, сухожилия, отдельные пучки. Исследование помогает обнаружить травмы, разрывы, посттравматические повреждения, образования.
Диагностика наследственных болезней мышц
Генетические исследования позволяют обнаружить мутации в известных генах, ассоциированных с миопатиями. Молекулярно-генетический анализ предоставляет возможность избежать дальнейших диагностических исследований и спрогнозировать течение заболевания. Благодаря генетической диагностике можно установить вероятность передачи болезней мышц по наследству.
Диагностика проводится с помощью таких методов, как:
- хромосомный микроматричный анализ (изучение всей структуры генома человека в одном исследовании позволяет исследовать последовательность генетического материала во всех хромосомах, обнаружить его избыток или недостаток);
- экзомное секвенирование нового поколения (заключается в анализе кодирующих последовательностей всех известных генов или специально отобранных генов, назначается при спорных клинических случаях).
| Наименование | Срок выполнения, дни | Цена исследования, руб |
| Панель "Нейродегенеративные заболевания" | 90 | 35000 |
| Панель "Наследственные нарушения обмена веществ" | 90 | 35000 |
| Панель "Наследственные эпилепсии" | 90 | 35000 |
| Панель "Нервно-мышечные заболевания" | 90 | 35000 |
| Панель "Умственная отсталость и расстройства аутистического спектра" | 90 | 35000 |
По результатам диагностики болезней мышц рекомендуется получить консультацию невролога и генетика.



Нервно-мышечные заболевания (НМЗ) - обширная группа заболеваний, характеризующихся прогрессирующим вялым парезом или параличем различных мышечных групп, в результате поражения определенных отделов рефлекторной дуги, обеспечивающей нормальную функцию мускулатуры.
Утрата или изменение нормальной двигательной активности из-за снижения силы и тонуса мышц может возникать как в результате поражения собственно мышц (первично-мышечные заболевания), так и в результате поражения периферических нервов (различные виды полинейропатий) или мотонейронов спинного мозга (спинальные мышечные атрофии), или дисфункции нервно-мышечного соединения (миастении), а также нарушений, косвенно влияющих на работу мышц.
Основными причинами возникновения НМЗ считаются аутоиммунные болезни, наследственные/генетические факторы и отравление различными веществами. Также в эту группу относятся часть врожденных дефектов метаболизма (в том числе болезни накопления и митохондриальные заболевания) и нейродегенеративных заболеваний со схожими симптомами.
- Гипер-калимический периодический паралич
- Гипo-калимический периодический паралич
- Дистальная мышечная дистрофия
- Болезнь Помпе
- Боковой амиотро-фический склероз
- Врожденная парамиотония
- Периодический паралич
- Миопатия с гигантскими митохондриями
- Дефицит рецепторов к ацетилхолину
- Болезнь Шарко-Мари-Тус
- Дефицит аденилат-дезаминазы
- Врожденная миопатия
- Дефицит карнитина
- Мышечная дистрофия Эмери-Дрейфуса
- Врожденная миастенические синдромы
- Болезнь центрального стержня
- Поясно-конечностная мышечная дистрофия
- Центрально-ядерная миопатия
- Мито-хондриальная миопатия
- Немалиновая миопатия
- Болезнь Помпе
- Дистальная миопатия
- Спинальная амиотрофия
- Мышечная дистрофия Беккера
- Врожденная мышечная дистрофия
- Мышечная дистрофия Дюшенна
- Лице-плече-лопаточная мышечная дистрофия
- Синдром Гийена-Барре
- Миотоническая дистрофия
- Миозит
- Окуло-фарингеальная мышечная дистрофия
Наследственные нервно-мышечные заболевания (ННМЗ)
Наиболее распространенная группа моногенных болезней нервной системы с различными типами наследования (аутосомно-доминантным, аутосомно-рецессивным, х-сцепленным, материнским) и практически во всех ранее обследованных территориях России и других популяций мира оставляет более 50% в структуре нозологического спектра наследственных болезней нервной системы.
Значительное сходство клинических проявлений в группе ННМЗ (симптомы вялого паралича различных мышечных групп) создает значительные трудности при проведении их дифференциальной диагностики на этапе клинического обследования.
Неправильно поставленный топический диагноз приводит к ошибкам в диагностике определенной нозологической формы ННМЗ, что увеличивает экономические затраты на проведение дорогостоящих молекулярно-генетических анализов и/или не позволяет осуществить профилактику возникновения повторных случаев заболевания в отягощенных семьях, определять генетический статус родственников больного, а также проводить подбор адекватной патогенетической терапии возможнойв некоторых случаях заболеваний этой группы.
| Неврология и нейрохирургия Форумы: Форум для общения врачей неврологов и нейрохирургов, Мануальная терапия |
| Поиск по форуму |
| Расширенный поиск |
| Найти все сообщения с благодарностями |
| Поиск по дневникам |
| Расширенный поиск |
| К странице. |
 | 1.jpg (327.1 Кб, 73 просмотров) |
| 2.jpg (230.0 Кб, 59 просмотров) |
Ситуация ваша не для заочного консультирования.
Надеюсь, у вас есть на руках стекла, чтобы можно было бы провести независимую оценку морфологических изменений биоптата? Это важно, т.к. стороннее экспертное мнение необходимо.
Данные биопсии кожно-мышечного лоскута должен интерпретировать ревматолог, специализирующийся на васкулитах.
В целом, всё же, по совокупности ваших жалоб и данных на мой взгляд фибромиалгия более вероятна.
Креатинкиназа 54
Из огромной кучи исследований не очень хорошие только ЭТИ:
Кортизол 530 (171-536)
ТТГ 6,48 (0,17-4,0)
УЗИ диффузные изменения пожделудочной, деформация формы желчного пузыря
Холестерин примерно такой лет 20 или больше:
Холестерин общ 7,22 (2-5)
Холестерин ЛПНП 4,94(2,51-3)
Триглицериды 1,98 (0,3-1,7)
Индекс атерогенности 5,27 (0,0-4,88)
Суточный уринолизис Креатин -2,7 (0) Проба Фелинга Фиолетовая
Все 5 лет болезни
Нейтрофилы 41,2 (47-72)
Лимфоциты 46,3 (19-37)
HLAB-27 отрицательно 2 года назад и положительно в этом году
Есть некоторые проблемы ЖКТ (с 7 лет ГЭРБ)
Неделю назад выписался из Обл.Неврологии 2(генетика) после 3 недель обследований - ничего не поймали, в выписке Миопатический синдром (назначили Тиоктовую и Глутаминовую кислоту). После выписки вообще сидеть более 2 часов не могу (за рабочим местом и дома) - шея дубеет, грудной отдел позвоночника тоже (как будто мерзнет), но на наклоны и повороты болей нет - просто мышцы спины как говорят спортсмены "забиты". Обострения к вечеру (стынут еще и конечности). Объективно мышцы не терпят нагрузку (18-20 раз имитация в пустом кулаке выжим гантели и тремор и больше 25 рука не идет вверх; прижать банку при закатке консервов дома не могу - тремор через 20 секунд и тд ) А еще ощущение в мышцах почти постоянное как у неопытных спортсменов перед стартом (похоже на адреналин, но почти не связано с нервным возбуждением, хотя раньше было только при переживаниях). Плохо еще и то, что грудные мышцы вовлекаются в эти ощущения сильно. Доктора без протокола почти все прямым текстом - ищи поддержку по телемедицине, стучись в НИИ Неврологии. А, да, в ПН собрав круглую сумму иду сдавать кровь на панель нервно-мышечных заболеваний с доставкой в Геномед Москва. Через 90 дней если дотяну может что и узнаю :-(
| в ПН собрав круглую сумму иду сдавать кровь на панель нервно-мышечных заболеваний с доставкой в Геномед |
Не обижайтесь, и спасибо за сочувствие (просто договор и предоплата ушла им безналом еще неделю назад, а сдать по платежке в Омске в их лабе-партнере уже дело было неотвратимое).
Скорее всего так и есть - местная ОКБ "заземлила" меня на Геномед от отсутствия собственных версий и нежелания говорить "мы не знаем".
Уважаемые доктора, решил реанимировать свою тему - накопилось много новых симптомов, а диагноза так и нет. Между последними сообщениями в теме и сегодняшним днем прошло почти 4 года. Были и плановые госпитализации и посещения неврологов и даже поездки в Москву в НИИ неврологии (откуда отправили к весьма известному профессору по нервно-мышечным патологиям). Панель нервно-мышечных заболеваний Геномеда ничего не нашла. Везде посочувствовали и напутствовали жить дальше по принципу "привезут - будем лечить, а пока ходишь-ходи" или "плохой стук себя проявит". А между тем изменения есть (хотя и не тянут на неотложку :-) ).
1. При посещении стоматолога не могу держать открытым рот более 20 минут (а надо иногда и час!) - сперва челюсть, потом голову начинает трясти. Врачи пугаются, да и работать не могут.
2. Стал замечать тремор намерения. В общем случае возникает в неудобной позе (как бы мышцы быстро устают и начинают сокращаться). Но иногда возникают в момент намерения совершить какое-либо движение.
3. Довольно быстро устает спина в любой статической позе (мытье посуды, готовка на кухне и тд - часто за 3-5 минут)
4. Стали затекать от любой слегка неудобной позы конечности. Может это нормально, но не за 5-7 минут же. В целом же как и прежде ворочаюсь ночь напролет - меняю положение тела.
5. Часто холодеют конечности. Холодно. Но в течение дня могут вдруг наступить самопроизвольно неожиданные и необъяснимые короткие моменты "оттепели".
6. При работе за компьютером в полусогнутых локтях иногда возникают болезненные ощущения (ну как бывает неудачно стукнешься локтем и дикая боль - только тут умеренная и без удара; я о том, что она не суставного происхождения)
7. Иногда от нескольких часов до нескольких дней возникает подергивания разных мышц (бедра, кистей, спины)
8. Подъем по ступенькам на 7-9 этаж совершить могу, а вот обратно спускаться уже проблема - ноги странно подкашиваются.
9. Стал часто ловить себя на том, что держу зубы стиснутыми (иногда корренные плотно сжаты, но чаще передние нижние прижаты до тупой боли к верхним изнутри)
Как и прежде боли и спазмы мышц щеи удается ненадолго снять неким подобием самомассажа (продавливание большим пальцем шеи в разных направлениях вплоть до сильной боли потом на несколько минут (может и десятков) приводит к непродолжительному облегчению)
Как и прежде напряжение ненадолго снимает прогрев в горячем душе.
На несколько часов снимает спазм мышц спины и часовое плавание кролем. Или посещение сауны, но ненадолго - до остывания.
Готов по вашим рекомендациям сдать любые (повторные) анализы. Хотя наверно с посещением врачей в клиниках не все так просто теперь. Буду благодарен за любые мнения. Если идеи не очень традиционные (и не корректно выносить их на публику) для темы, можно в личку.
Нервно-мышечные заболевания характеризуются нарушением функции произвольной мускулатуры, утраты или снижения двигательного контроля, которое может наступать в результате поражения как собственно мышц, так и иметь вторичный характер – вследствие дисфункции нервно-мышечного соединения, поражения периферических нервов или мотонейронов спинного мозга. В клинической картине некоторых нервно-мышечных заболеваний могут присутствовать признаки поражения двигательных ядер ствола головного мозга. Поражения других участков нервной системы, приводящих к нарушению двигательного контроля, в частности пирамидного тракта, согласно общепринятому определению не относятся к нервно-мышечным заболеваниям.

Наиболее частыми симптомами нервно-мышечных заболеваний являются слабость, снижение мышечного объема (атрофия), непроизвольные мышечные подергивания, спазмы, онемение, покалывание и др. Нарушение функции нервно-мышечного соединения может вызывать опущение век (птоз), двоение в глазах (диплопия), и другие признаки мышечной слабости, которые усиливаются в течение дня. При некоторых заболеваниях могут нарушаться глотание, и даже дыхание.
Заболевания мышц: симптомы
Заболевания нервно-мышечного соединения
Вызывают дисфункцию нормальной синаптической передачи импульсов с нервных окончаний на мышечные волокна. В основе заболевания может лежать аутоиммунный процесс.
- миастения гравис
- синдром Ламберта-Итона
- врожденный миастенический синдром
Заболевания периферических нервов
- Мононейропатии - поражение одного нерва, наиболее частой причиной является компрессионное воздействие (туннельные синдромы), травматические повреждения
- Множественные мононейропатии - мультифокальные поражения периферических нервов, связаны обычно с системными или инфекционными заболеваниями, паранеопластическими синдромами
- Полинейропатии - диффузные, симметричные поражения периферических нервов, обычно преобладаюшие в дистальных отделах, почти всегда в клинической картине присутствуют расстройства чувствительности . Могут быть острыми (синдром Гиейна-Барре), хроническими (хроническая воспалительная демиелинизируюшая полинейропатия), приобретенными (токсические, диабетические, паранеопластические) или наследственными (перонеальная мышечная атрофия или болезнь Шарко-Мари-Тус, болезнь Дежерина-Сотта, атаксия Фридрейха).
- Плексопатии - поражения сплетений верхних и нижних конечностей (плечевого и пояснично-крестцового), наиболее частой причиной которых являются травматическое или компрессионное воздействие
- Радикулопатии и полирадикулопатии - поражения двигательных или чувствительных спинальных корешков
Заболевания двигательного нейрона
Прогрессирующее дегенеративное поражение мотонейронов, которое наиболее заметно приводит к нарушению двигательного контроля верхних или нижних конечностей, а также бульбарным расстройствам. Чаще начинаются в среднем возрасте, симптомы могут включать слабость в конечностях, нарушение глотания, речи, походки, слабость лицевой мускулатуры, мышечные спазмы. В эту группу входят, в частности:
- боковой амиотрофический склероз (БАС)
- спинальная мышечная атрофия взрослых
- спинальная мышечная атрофия у младенцев или болезнь Верднига-Гоффмана
- юношеская спинальная мышечная атрофия или болезнь Кугельберга-Веландера
- бульбоспинальная мышечная атрофия или болезнь Кеннеди
Диагноз ставится на основе истории заболевания, тщательного неврологического осмотра, в большинстве случаев используется электромиографическое (ЭМГ) исследование, при сочетании с поражением центрального мотонейрона или для исключения его может применяться транскраниальная магнитная стимуляция, при подозрении на наследственные формы проводится анализ ДНК, аутоимунный характер процесса требует определения специфических антител, может проводиться биопсия участка мышцы, при первично-мышечных поражениях проверяется уровень содержания креатинфосфокиназы (КФК), в последнее время набирает также популярность ультразвуковое исследования мышц и периферических нервов. Диагностический алгоритм, выбор дополнительных исследований зависят от особенностей клинического паттерна и локализации поражения - мышца, нерв, сплетения, корешки, двигательные нейроны.

Нервно-мышечные заболевания (НМЗ) – что это такое?
В медицине под этим термином подразумевается большая группа болезней, передающихся на генетическом уровне от родителей к ребенку.
Им характерно нарушение функций мускулатуры, низкая двигательная активность или ее отсутствие.
Такие последствия наступают в результате нарушений функций нервно-мышечных соединений, поражения мышц, спинномозговых нейронов или нервов.
Основные причины возникновения НМЗ – это аутоиммунные болезни, отравление различного рода веществами и наследственный фактор. Также сюда стоит отнести часть врожденных дефектов метаболизма.
Классификация
К НМЗ относятся:
- первичные прогрессирующие мышечные дистрофии (миопатии);
- вторичные прогрессирующие мышечные дистрофии;
- врожденные непрогрессирующие миопатии;
- миотонии;
- наследственныепароксизмальные миоплегии.
О каждом из видов поговорим подробнее дальше.
Первичные прогрессирующие мышечные дистрофии (миопатии)

К признакам прогрессирующих мышечных дистрофий (ПМД) относятся дегенеративные изменения в мышцах.
Они заключаются в истончении мускулатуры, замене мышечной ткани на жировую и соединительную.
Возникает фокальный некроз, поперечная исчерченность теряется.
Некоторые механизмы возникновения и течения болезни до сих пор не установлены. Причиной миопатии являются мембранные дефекты в мышечных клетках. Большие надежды на установление деталей патогенеза возлагают на микробиологов.
При миопатиях мышцы слабеют, затем атрофируются. Существует несколько форм этой болезни. Разные виды ПМД различают по типу наследования, срокам проявления, характеру и течению болезни, расположением атрофий.
Вторичные прогрессирующие мышечные дистрофии
При вторичных дистрофиях (спиральных и невральных амиотрофиях) поражаются спинномозговые мононейроны и периферические двигательные нервы. Это приводит к вторичному поражению мышц и их атрофии.
Болезни этого типа прогрессируют. При изучении мышц больного обнаруживаются как нормальная, так атрофированная и гипертрофированная мышечная ткань.
Наследственные пароксизмальные миоплегии
Пароксизмальные миоплегии – редко встречающиеся наследственные заболевания у детей. Характеризуется периодическими приступами паралича мышц скелета. Наследуется по принципу, при котором для проявления болезни достаточно одного мутантного аллеля, локализованного в аутосоме.
Выделяют три формы:
- гиперкалиемическая;
- гипокалиемическая;
- нормокалиемическая.

Гипокалиемической формой пароксизмальной миоплегии чаще всего болеют мужчины.
Первые приступы заболевания появляются с 10 лет.
Приступ возникает в утренние или ночные часы. При этом чувствуется слабость в конечностях, шее, часто доходящая до паралича. В отдельных случаях паралич распространяется на лицевую мускулатуру и дыхательные пути.
Приступ сопровождается жаждой гиперемией, потливостью. Длительность может составлять от часа до семи дней. Женщины обычно испытывают приступ в первый день менструаций.
Гиперкалиемическую форму можно встретить реже, ее приступы начинаются в младшем возрасте. Приступ провоцируют холод и длительное бездействие мышц. Он начинается с расстройства чувствительности лицевых мышц, рук и ног.
Третья форма болезни проявляется до 10-летнего возраста. Приступы проходят в течение нескольких дней или недель. Они могут быть спровоцированы низкой температурой окружающей среды, употреблением алкоголя, большими физическими нагрузками.
Справка! Наследственные пароксизмальные миоплегии могут быть симптомами болезней щитовидной железы, Адиссона, реже встречаются при рвоте и поносе.
Врожденные непрогрессирующие миопатии
Непрогрессирующая миопатия характеризуется доброкачественностью, проявляется в младенчестве или в детстве. К этой разновидности НМЗ относятся болезни центрального стержня, намалиновые и митохондриальные миопатии. При этом возникают метаболические мышечные изменения. Пациент чувствует снижение сил, ослабление рефлексов.
Миотонии

Миотония – это замедленное расслабление мышцы после ее напряжения.
Болезнь разделяют на три вида:
- миотония действия;
- перкуссионная миотония;
- электромиографическая миотония.
Если больной с миотонией действия сожмет пальцы в кулак, а затем попытается быстро их разогнуть, то ему понадобится время, чтобы полностью выпрямить ладонь.
Вторая разновидность отличается сокращением мышц при интенсивном постукивании молоточком.
Чтобы обнаружить электромиографическую миотонию, потребуется игольчатый разряд. При этом аппарат показывает разряды высокой частоты, которые сначала учащенные, а потом снижают частоту.
Список всех НМЗ
- Болезни ЦС;
- Миотонии Томсена;
- Болезни Штейнерта-Куршманна;
- Миодистрофии Беккера;
- Миодистрофии Дюшенна;
- Миодистрофии Ландузи-Дежерина;
- Митохондриальные миопатии;
- Невральные амиотрофии Шарко-Мари-Тута;
- Немалиновые миопатии;
- Офталъмоплегические миопатии;
- Спинальные амиотрофии Верднига-Гоффманна;
- Спинальные амиотрофии Кугельберга-Веландер;
- Врожденная мышечная дистрофия;
- Мышечная дистрофия Беккера;
- Болезнь Помпе;
- Миотоническая дистрофия;
- Мышечная дистрофия Дюшенна.
Симптомы
Определяющий симптом НМЗ – это слабость в мышцах. Зонами поражения являются плечевой пояс, бедра, таз, область плеч. При каждом виде болезни поражается конкретная группа мышц. Мышцы поражаются симметрично.
Больной задействует другие мышцы, чтобы справиться с определенной задачей. Например, если поражена область ног, то, чтобы подняться с пола, человек сначала опирается руками, встает на колени, берется за опору, а потом уже может сесть на кресло. Сесть, не используя руки, он не может.
Справка! Редко встречаются случаи функциональных нарушений лицевых мышц. При этом возникает опущение века, губы, появляются речевые нарушения, трудно глотать.
Большинство заболеваний проходят с одинаковыми признаками. Со временем мышца атрофируется, возникает псевдогипертрофия из-за разрастания соединительной ткани. Возникает боль, подвижность в суставе ограничивается.
Диагностика
В диагностике нервно-мышечных заболеваний важны следующие методы:
- биохимические;
- электрофизиологические;
- патоморфологические;
- ДНК-диагностики.
Биохимический метод заключаются в том, что у больного определяют ферменты мышц.
Важно! Повышение уровня ферментов может быть вызвано и у здоровых людей при физической нагрузке.
При разных видах НПЗ уровень фермента повышается в разное количество раз по сравнению с нормой. Это позволяет определить форму заболевания.
Электрофизиологический метод включает в себя электронейромиографию в сочетании с электромиографией.
Патоморфологический метод включает мышечную биопсию – изучение мышцы под световым микроскопом.
Наиболее эффективным методом определения болезни является ДНК-диагностика. Полимеразная реакция позволяет выявить НПЗ в 70% случаев.
Способы лечения

Лечение направлено на поддержку мышечных сил, замедление атрофирования.
Цель терапии заключается в том, чтобы пациент как можно дольше передвигался без посторонней помощи, т.к. в постоянном вертикальном положении возникают расстройства дыхания.
Основными способами лечения являются:
- Лечебно-физкультурный комплекс. Это различные активные и пассивные движения. Нельзя подвергать больного усиленной нагрузке. Упражнения должны быть регулярными;
- Ортопедические мероприятия. Применение специальных шин и проведение операций. Эти меры направлены на сохранение самостоятельных передвижений;
- Лекарственные препараты. Медикаменты, поддерживающие обмен веществ, устраняющие дефицит энергии и белка. Больным назначают фосфаден, преднизолон, нифедипин, витамин Е.
Неправильно поставленный предварительный диагноз приводит к ошибкам в диагностике формы НМЗ. Это увеличивает траты на осуществление дорогостоящих анализов и препятствует осуществлению профилактики рецидива, определения генетического статуса предков пациента и подбора адекватного патогенетического лечения, которым можно ограничиться в некоторых случаях.
Полезное видео по теме:
Читайте также:
