
Нервно мышечные заболевания шарко мари
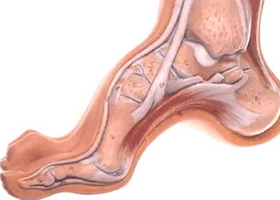
Общие сведения
Невральная амиотрофия Шарко Мари Тута объединяет группу наследственных прогрессирующих хронически протекающих полиневропатий:
- болезнь Рефсума;
- гипертрофическая невропатия Дежерина-Сотта;
- синдром Русси-Леви.
Частота встречаемости варьирует от 2 до 36 случаев на 100 тысяч населения. Невральная амиотрофия носит в основном семейный характер и у разных родственников клиническая симптоматика может сильно разниться. Параллельно регистрируются спорадические варианты течения болезни Шарко-Мари-Тута. Невральная амиотрофия чаще поражает лиц мужского пола.
Болезнь Шарко Мари Тута относится сразу к нескольким заболеваниям, которые получили название от имени Жана-Мартина Шарко:
Патогенез
Большинство форм болезни связано с поражением миелиновых оболочек в волокнах периферических нервов. Гораздо реже встречается патология аксонов (осевые цилиндры, которые проходят по центру нервного волокна). Дегенеративные изменения наблюдаются также в путях Голля (проводящая система глубокой чувствительности в спинном мозге), нейронах передних рогов, корешках спинного мозга (передние и задние), столбах Кларка (относятся к заднему спинномозжечковому пути).
На фоне дисфункции периферической нервной системы развивается мышечная атрофия, которая затрагивает отдельные группы миофибрилл. По мере прогрессирования патологии происходит смещение ядер сарколеммы, интерстициальное разрастание соединительной ткани, поражение миофибрилл. При нарастании гиалиновой дегенерации миофибрилл наблюдается их распад.
Классификация
Выделяют 2 типа невральной амиотрофии Шарко. Разграничение основывается на ряде особенностей, но в целом клиническая симптоматика у обоих типов схожа.
- I тип. Характерно выраженное снижение скорости проведения нервных импульсов. При биопсии нерва выявляется сегментарная демиелинизация нервных волокон и гипертрофическое разрастание непоражённых шванновских клеток.
- II тип. Скорость проведения импульсов практически не страдает, а при анализе биоптата выявляется дегенерация аксонов.
Также есть связь между атаксией Фридрейха и болезнью Шарко-Мари-Тута. У некоторых пациентов с ШМТ диагностируется клиническая симптоматика атаксии Фридрейха и наоборот (в течением времени). Также встречаются промежуточные формы этих заболеваний. В медицинской практике описаны случаи выявления атаксии Фридрейха у одних родственников и амиотрофии ШМТ – у других.
Причины
Достоверных данных о причинах развития невральной амиотрофии на сегодняшний день в практической неврологии нет. У 70-80% пациентов с болезнью Шарко отмечалось дублирование 1 участка 17 хромосомы. У патологии есть несколько форм, что обусловлено мутациями различных генов. Для заболевания характерен аутосомно-доминантный тип наследования с показателем пенетрантности на уровне 83%. Также были зарегистрированы случаи аутосомно-рецессивного типа наследования.
Симптомы болезни Шарко
В некоторых случаях заболевание начинается с нарушения чувствительности в стопах, и очень часто – с парестезий (чувство ползания мурашек по стопам). Характерным ранним признаком заболевания является отсутствие сначала ахилловых, а затем и сухожильных, коленных рефлексов. Часто пациенты предъявляют жалобы на болезненные, приступообразные сокращения в икроножных мышцах, которые усиливаются после длительной физической активности либо в ночное время.
Далее болезнь прогрессирует и поражает дистальные отделы рук – сначала кисти, а затем атрофия касается и мышц предплечий. Кисть становится похожей на лапу обезьяны из-за поражения тенара и гипотенара. Атрофические изменения никогда не регистрируются в мышцах плечевого пояса, туловища и шеи.
Очень часто отмечаются лёгкие фасцикулярные подёргивания мышц рук и ног. В некоторых случаях появляется компенсаторная гипертрофия в мышцах проксимальных отделов конечностей. Чувствительные нарушения проявляются тотальной гиперстезией. Болевая и температурная чувствительность страдают больше, чем глубокая. Редко появляется отёчность в поражённых конечностях и цианоз кожных покровов.
Клинические симптомы при болезни ШМТ прогрессируют очень медленно. Временной промежуток между поражением верхнего и нижнего пояса может достигать 10 лет. Длительное время пациенты остаются трудоспособными. Негативное влияние на скорость развития патологического процесса могут оказывать разные экзогенные факторы:
- черепно-мозговая травма;
- переохлаждение;
- перенесённые инфекции (краснуха, мононуклеоз, корь, ОРВИ, ангина);
- гиповитаминоз;
- травма позвоночника, спинного мозга.
Анализы и диагностика
Обследованием пациентов с подозрением на Болезнь Шарко-Мари-Тута занимаются невропатологи и ортопеды. При первичном осмотре уточняется возраст пациента, в котором впервые стала появляться характерная симптоматика. Обязателен сбор семейного анамнеза, с уточнением наличия у родственников схожих генетических заболеваний. При осмотре врач обращает внимание на деформацию кистей рук и стоп ног, на изменение походки.
Во время неврологического осмотра выявляется снижение тонуса в дистальных отделах верхних и нижних конечностей, снижение чувствительности кожных покровов и ослабление (вплоть до полного отсутствия) сухожильных рефлексов (коленные, ахилловы).
Основные методы исследования:
Дифференциальная диагностика проводится с заболеваниями:
- синдром Гийена-Барре;
- адренолейкодистрофия;
- спинальная мышечная атрофия Верднига-Гоффмана;
- болезнь Пелицеуса-Мерцбахера.
Лечение
Терапия пациентов с болезнью Шато проводится в стационарных условиях. Какого-либо специфического лечения нет, чтобы замедлить прогрессирование демиелинизации и аксональной дегенерации. Индивидуальная, грамотно подобранная терапия позволяет улучшить качество жизни пациента.
Частота диагностирования болезни Шарко-Мари-Тута согласно статистическим данным где-то один случай на две с половиною тысячи пациентов. Первые симптомы появляются в молодом возрасте. Выраженность симптомов и скорость прогрессирования болезни Шарко-Мари разная у каждого пациента. Процент инвалидизации при заболевании очень высок.

Причины заболевания болезнью Шарко:
- Мутация гена PMP22;
- Мутация MPZ;
- Мутация GJB1;
- Мутация MFN и др.
- аутосомно-доминантный (чаще всего);
- аутосомно-рецессивный;
- Х-сцепленный.
Заболевание имеет много форм, вызванные разным видом мутаций. Качество жизни и возможности работоспособности при болезни Шарко-Мари-Тута значительно ухудшаются, но на продолжительности жизни обычно это не сказывается.
Симптомы болезни Шарко связанные с поражением моторных и сенсорных нервных волокон. Диагностика болезни Шарко-Мари заключается в исключении диагнозов, которые могут давать подобную клиническую картину, и в проведении ДНК-диагностики, но учитывая, что не все виды мутаций известны, она не всегда информативна.
Лечение болезни Шарко-Мари-Тута заключается в симптоматической терапии. Специфическое лечение на данный момент все еще находится в стадии разработки.
Юсуповская больница – одно из лучших медицинских учреждений, где лечатся пациенты с болезнью Шарко-Мари-Тута. Несмотря на то, что терапия направлена только на купирование симптомов, неврология стремительно развивается и питается найти способы лечения многих заболеваний, в том числе и болезни Шарко. Ведение пациентов изданной патологией достаточно сложное, ведь клиническая картина разнообразна, симптомы выражены в неодинаковой степени и п.т. Опыт работы специалистов Юсуповской больницы позволяет оказывать качественную и эффективную медицинскую помощь. Доктора следят за клиническими исследованиями, новыми разработками, препаратами, изучают их эффективность. В случае необходимости, диагностика проводится быстро и с использованием нового оборудования. Персонал работает на благо пациента.
Симптомы болезни Шарко
Симптомы болезни Шарка-Мари появляются в молодом возрасте, чаще всего до двадцати лет. Прогрессирует заболевание постепенно, пациенты долгое время сохраняют работоспособность и возможность самообслуживания. Причинами, которые ускоряют развитие заболевания, могут стать вирусные и бактериальные инфекции, воздействия неблагоприятных факторов среды, травматизм, недостаток витаминов и т.п.
Дальнейшее развитие заболевания болезни Шарко-Мари-Тута приводит к втягиванию в процесс и кистей, далее - предплечий. Мышцы шеи, туловища и плечевого пояса не атрофируются. Мышцы проксимальных отделов компенсаторно увеличиваются.
Все виды чувствительности нарушаются, но больше всего страдает поверхностная, особенно температурная.
Диагностика болезни Шарко
- Осмотр неврологом;
- Лабораторные исследования;
- Инструментальные исследования;
- ДНК-исследования.
Полное обследование необходимо для исключения заболеваний, в которых клиническая картина сходна. К ним относят: боковой амиосклероз, миотония, метаболическая невропатия. Для исключения хронических полинейропатий проводят биопсию мышц.
Лечение болезни Шарко
Все способы лечения болезни Шарко-Мари-Тута не радикальны. Симптоматическое лечение включает медикаментозную терапию, физиотерапию, лечение у ортопеда и т.п.
Физиотерапевтическое лечение болезни Шарко-Мари-Тута включает ЛФК, массаж, электрофорез, диадинамотерапию, терапию лечебными грязями, разные виды ванн и др.
Медикаментозная терапия направлена на улучшение питания мышечных волокон. С этой целью назначают кокарбоксилазу, глюкозу, аденозинтрифосфат и др. Так же широко применяют антиоксидантные средства, препараты, улучшающие микроциркуляцию, и витамины. Хорошо зарекомендовали себя препараты, которые тормозят активность ацетилхолинэстеразы и повышают уровень ацетилхолина, например, прозерин, галантамин.
Дальнейшие разработки новых препаратов, направленные на радикальные меры – это мир без болезни Шарко-Мари-Тута. Прогрессирование болезни Шарко-Мари-Тута не отражается на том, сколько живут пациенты.
В Юсуповской больнице специалисты долгие годы помогают пациентам держать болезнь под контролем. Минимальная выраженность симптомов и медленное прогрессирование – результат работы врачей. В комфортных палатах, на новых тренажерах, в хорошо оснащенных кабинетах – вот где проходит лечение болезни Шарко-Мари-Тута. Не затягивайте с лечением, запишитесь на консультацию.
Синдром Шарко считается одной из самых распространенных форм наследственных заболеваний, характеризующихся поражением нервных тканей. Болезнь составляет примерно 80% от неврозов генетического характера.

Характерные черты патологии были описаны в конце 19-го века тремя врачами: французами Жаном-Мартином Шарко, Пьером Мари и англичанином Говардом Генри Тутом. В их честь и появилось название – болезнь Шарко-Мари-Тута.
Используемые синонимы – наследственная моторно-сенсорная невропатия, невральная амиотрофия. Связана патология с воздействием на периферические нервы, вследствие чего происходит разрушение миелиновой оболочки или длинных отростков нервов – аксонов.
Принято считать, что при этом заболевании не происходит поражение ЦНС. Однако есть данные, свидетельствующие о том, что разрушение затрагивает корешки спинного мозга, проводниковые пути.
Вследствие нарушения проводимости нервных волокон, расположенных на периферии, атрофируются мышечные ткани конечностей. Постепенно происходит их замена соединительными и жировыми тканями.
Симптомы болезни Шарко-Мари-Тута диагностируются чаще всего у детей и молодых людей от десяти до двадцати лет.
Причины
Нервные импульсы передаются по длинным отросткам нейронов — аксонам. Они обернуты миелиновой оболочкой. В ее создании принимают участие олигодендроциты. При патологии Шарко-Мари-Тута мутация происходит в гене MFN2. В зоне его ответственности – выработка митохондрального белка. Мутация ведет к формированию сгущений митохондрий в теле аксона.
Вероятно, заболевание вызвано еще и воздействием генов на иммунную систему организма. В результате белки миелинового полотна начинают восприниматься как свойственные патологическим бактериям. Активизируется иммунная система, образуются антитела, они проникают сквозь гемато-энцефалический барьер и поражают белковые компоненты.
Вследствие воздействия некоторых генов происходит чрезмерная миелинизация нервных клеток, что также нарушает прохождение нервных импульсов.
В патогенезе синдрома Шарко-Мари-Тута, таким образом, выделяют 2 формы:
- Обусловленную разрушением миелиновой оболочки. Диагностируется примерно в 80% случаев.
- В основе второй лежит поражение аксонов. Встречается значительно реже.
Передача патологии осуществляется преимущественно аутосомно-доминантным путем, т. е. ребенок получает ее от одного из родителей. В некоторых случаях отмечается рецессивная передача – оба родителя являются носителями патологических генов, а для развития болезни нужны две генных копии.
Редко возникает мутация гена у одного человека, не связанная с наследственными факторами. Точные причины патологии до настоящего времени неизвестны.
Симптомы
Синдром Шарко ведет к повреждению двигательных и чувствительных нервов. Разрушение моторных путей сопровождается слабостью и онемением мышечной ткани обеих стоп, быстрым нарастанием усталости. Несколько позже присоединяются болевые ощущения в икроножных мышцах. Развиваются они преимущественно после долгой ходьбы, стояния на одном месте.

Во время осмотра обнаруживается атрофия мышечных волокон ног. Угнетаются сухожильные рефлексы.

Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
МКБ-10

- Причины
- Патогенез
- Классификация
- Симптомы
- Осложнения
- Диагностика
- Лечение невральной амиотрофии Шарко-Мари-Тута
- Медикаментозная терапия
- Немедикаментозная терапия
- Хирургическое лечение
- Экспериментальное лечение
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание – приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Диагностика
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
- ЭНМГ. При электронейромиографии отмечаются признаки аксональной и демиелинизирующей нейропатии – замедление скорости проведения импульса по двигательным нервам, падение амплитуды М-ответов.
- Компьютерная паллестезиометрия. Данная диагностическая процедура позволяет объективно оценить снижение вибрационной чувствительности – наиболее ранний признаки болезни ШМТ.
- Гистология. При гистологическом исследовании биоптата большеберцового нерва обнаруживаются уменьшение количества миелиновых волокон, разрастание соединительнотканных волокон, атрофию миелина.
- ДНК-анализ. Подтверждающий метод исследования, верифицирующий диагноз. Выявляются дупликации гена белка периферического миелина (PMP22) на 17-й хромосоме.
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру – баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Комплексное проведение данных мероприятий позволяет увеличить мышечную силу, исправить нарушения равновесия и походки. Благодаря этому удается повысить бытовую, социальную адаптацию, работоспособность пациентов.
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства – метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 – ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута – тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Болезнь Шарко-Мари-Тута характеризуется поражением периферических отделов нервной системы, проявляется атрофическими изменениями в мышцах конечностей. Чаще диагностируется у мужчин в возрасте 15-30 лет. Первые признаки включают слабость в зоне стоп, быструю утомляемость ног при ходьбе. Пациентам сложно долго пребывать в положении стоя, поэтому они начинают перетаптываться на одном месте, чтобы уменьшить мышечное напряжение в ногах.
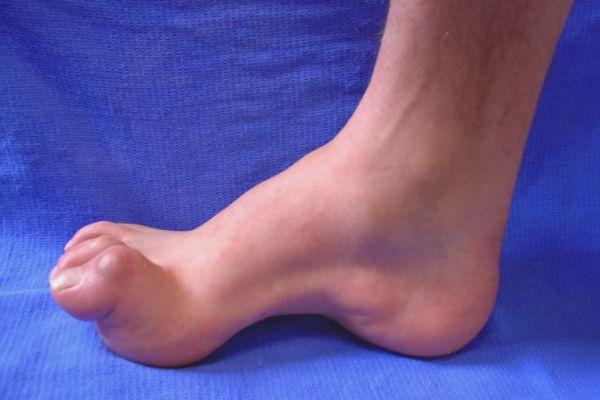
Характеристика
Невральная амиотрофия, известная так же как болезнь Шарко-Мари-Тута, является генетическим заболеванием, которое проявляется ослаблением и истончением мышц голени. Атрофические изменения в мышечной ткани приводят к уменьшению объема мускулатуры в зоне нижних конечностей.
Болезнь Шарко-Мари-Тута характеризуется повреждением нервных окончаний, которые передают информацию от соматосенсорных рецепторов к отделам головного мозга. Одновременно происходит повреждение нейрональных моторных путей, по которым передаются сигналы от корковых центров управления исполнительным органам – скелетным мышцам.
Болезнь Шарко-Мари-Тута передается по наследству (чаще по аутосомно-доминантному типу), рассматривается как нейропатия (нарушение функций нервов) сенсорно-моторного типа. Дебют заболевания может случиться в раннем детском или более позднем возрасте. Распространенность патологии составляет 1 случай на 2500 человек.
Выделяют формы заболевания с учетом характера повреждения нервных окончаний, принадлежащих периферическому отделу нервной системы. Патология 1 типа развивается вследствие демиелинизации – разрушения миелиновой оболочки, покрывающей нервы. В норме миелиновая оболочка улучшает передачу нервных импульсов. Дебютирует чаще в среднем (4-5 лет) детском возрасте, реже в подростковом периоде.
Первые симптомы: слабость в стопах, атрофические изменения в мышцах дистальных отделов ног. Атрофические изменения в мышечной ткани кистей рук развиваются позже. Вместе с утратой чувствительности (неспособность различать температуру, воспринимать болевые раздражители, вибрацию) в зоне кистей рук и стоп ног наблюдается понижение (иногда выпадение) сухожильных рефлексов.
Полинейропатия Шарко-Мари-Тута 2 типа развивается на фоне повреждения аксонов – ответвлений нейронов, посредством которых осуществляется связь между отдельными элементами нейрональной сети. Это медленно прогрессирующая форма. Дебютные симптомы могут появиться у детей старшего возраста. В ходе инструментальной диагностики выявляются относительно нормальные показатели скорости проведения нервных импульсов.
Параллельно наблюдается уменьшение разницы между амплитудными значениями потенциалов действия (волна возбуждения) чувствительных нервных клеток. Другая особенность – полифазные (многофазные) потенциалы действия в области мышечных волокон. Биопсия показывает аксональную дегенерацию валлеровского типа – процесс разрушения участка аксона.
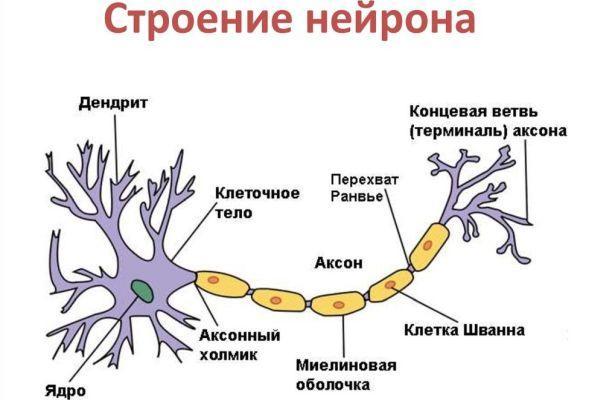
Причины возникновения и патогенез
Основная причина развития перонеальной атрофии 1 типа – дупликация (появление дополнительной копии участка хромосомы) гена PMP22 (17-я хромосома) миелинового белка в составе оболочки периферических нервов. В патогенезе заболевания Шарко-Мари-Тута лежит поражение двигательных нервных окончаний, что сопровождается нарушением регуляции сокращения мускулатуры.
В патогенезе участвует процесс повреждения сенсорных нервных окончаний, что приводит к нарушению передачи информации от рецепторов соматической чувствительности периферических отделов тела в соответствующие центры головного мозга. Невральная амиотрофия отражает дегенеративные, атрофические изменения в периферических нервных волокнах и реже в спинном мозге.
Симптоматика
Первый симптом Мари-Шарко-Тута – мышечная слабость в зоне стопы и голени (часть ноги от коленного сустава до пятки). Слабость в мышцах сопровождается изменением способности воспринимать внешние стимулы. Пациент теряет умение ощущать температуру внешней среды, вибрацию, боль. Прогрессирование заболевания сопровождается распространением потери чувствительности на проксимальные (расположенные ближе к центру тела) сегменты конечностей.
Клиническая картина существенно варьируется. У некоторых пациентов патология протекает бессимптомно, у других в ходе диагностического обследования выявляется уменьшение скорости проведения нервных импульсов в мышечной ткани, у третьих наблюдается значительное ограничение двигательной активности, связанное с мышечной атрофией и слабостью. Симптомы, типичные для болезни Шарко-Мари-Тута:
- Атрофические изменения в мышцах конечностей, преимущественно в ткани малоберцовых и дистальных (удаленных от центра, концевых) мышц. Из-за атрофии мышечной ткани голени нижние конечности напоминают ноги аиста или перевернутую вверх тормашками бутылку из-под шампанского.
- Мышечная слабость в области пораженных сегментов ног. Пациент испытывает трудности, если нужно согнуть ногу в области голеностопного сустава, чтобы в результате приподнялся дистальный сегмент стопы.
- Мышечная слабость в области пораженных сегментов рук. Самый поздний симптом, который указывает на прогрессирование патологии.
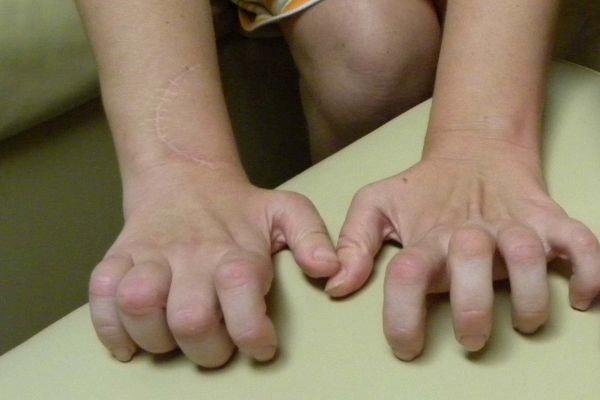
При легком течении могут наблюдаться единичные признаки, к примеру, своеобразная форма пальцев ног, которые напоминают молоток, или высокий подъем стопы. Обычно симптоматика у мужчин проявляется сильнее. У женщин часто симптомы отсутствуют. Синдром Шарко-Мари-Тута на начальных стадиях может включать признаки: болезненные ощущения острого характера в мышцах, парестезии (спонтанно возникающее чувство жжения, покалывания, ползания мурашек). Другие признаки БШМТ у детей и взрослых:
- Отвисание стопы. На фото пациентов отчетливо видно, что передняя часть стопы свисает вследствие ослабления мышц, если приподнять ногу. Больной не может самостоятельно поднять выше передний сегмент стопы.
- Нарушение походки, которая характеризуется как шаркающая, замедленная. Больной при ходьбе задевает носком поверхность, по которой идет. Чтобы предотвратить шарканье по поверхности, пациенты вынуждены высоко поднимать ноги при ходьбе.
- Болтающаяся стопа при значительном распространении атрофических процессов.
- Стопа Фридрейха (западание межпальцевых промежутков, высокий свод, пальцы в форме молотка).
Синдром Шарко-Мари-Тута включает симптомы: снижение чувствительности в дистальных сегментах конечностей, мышечные судороги в зоне предплечий и икроножных мышц. Типичный признак поздних стадий (спустя 10-15 лет после манифестации заболевания) – нарушение мелкой моторики рук, что сопровождается дисфункцией письма.
Пациент не может удерживать ручку, застегивать и расстегивать пуговицы на одежде. Реже наблюдаются вегетативно-трофические расстройства в виде гиперемии (покраснение кожных покровов) и повышенного потоотделения в области кистей и стоп. У пациентов часто диагностируются переломы костей стопы и растяжение связок, сухожилий в зоне лодыжки.
Диагностика
Диагноз ставят на основании клинических данных и сбора анамнеза. Чаще в семейном анамнезе упоминаются аналогичные случаи. Основной инструментальный метод диагностики заболевания Шарко-Мари-Тута – электромиография (исследование проводимости биоэлектрических импульсов мышечными структурами). Диагностические критерии:
- Мышечная слабость в дистальных отделах рук и ног.
- Ранний дебют заболевания (в детском, подростковом возрасте). Если патология дебютирует в младенческом возрасте, характерно тяжелое течение, которое часто приводит к инвалидизации.
- Наличие аналогичной симптоматики у близких родственников.
- Наличие деформаций в области пальцев и подъема стопы.
- Иногда болезненные ощущения при пальпации сосудисто-нервных пучков.
Подтверждением диагноза может служить результат генетического анализа, который указывает на наличие хромосомной мутации. В ходе нейровизуализации (МРТ, КТ) выявляются участки демиелинизации (ремиелинизации) в области периферических нервов. По результатам диагностического обследования врач назначает лечение заболевания Шарко-Мари-Тута.

Лечение
Болезнь Шарко Мари Тута лечится на основе разработанных клинических рекомендаций. Методов этиотропной (направленной на устранение причин заболевания) терапии не существует. Лечение болезни Шарко Мари Тута носит паллиативный характер, направлено на прекращение симптомов и улучшение качества жизни пациента. Лечение не может остановить прогрессирование невральной амиотрофии.
Однако использование ортезов (медицинские приспособления, предназначенные для изменения структурно-функциональных характеристик нервно-мышечного аппарата), физиопроцедуры, трудотерапия и психотерапия способствуют улучшению социальной адаптации и качества жизни больного. Обычно пациенты носят шины, поддерживающие свисающую стопу.
Иногда для стабилизации положения стопы проводится хирургическая операция. Лечение патологии Шарко-Мари-Тута предполагает регулярные, дозированные физические нагрузки и массаж, в том числе классический ручной, аппаратный и точечный. Лечебная гимнастика способствует уменьшению мышечной слабости и поддержанию двигательной активности.
Заболевание Шарко-Мари-Тута – наследственное, как и болезнь Виллебранда-Диана (наследственная патология крови, характеризующаяся возникновением спонтанных кровотечений по типу гемофилии), чтобы эффективно ее лечить, назначают комплексные мероприятия, такие как медикаментозная терапия, физиотерапия, бальнеотерапия (лечебные души и ванны), грязелечение.
Для улучшения трофики (питания) мышечной и нервной ткани, стимуляции обменных процессов назначают препараты: Кокарбоксилаза, АТФ, Рибоксин (участвует в метаболизме пуриновых нуклеотидов, необходимых для нормальных мышечных сокращений), Церебролизин (оказывает ноотропное действие), Метионин (регулирует липидный обмен).
Для улучшения микроциркуляции крови показаны препараты Пентоксифиллин, Никотиновая кислота. Коррекция проводимости импульсов в мышечной ткани осуществляется при помощи препарата Прозерин (ингибитор холинэстеразы). Комплексная программа медикаментозного лечения включает прием витаминов группы B, A, C, E.
Для профилактики деформации стоп носят удобную обувь, которая не сдавливает ногу и не стесняет движения. Нарушения походки корректируются подтяжками типа AFOs (ankle-footorthoses). Подобные приспособления помогают контролировать сгибание ног и голеностопного сустава, обеспечивают поддержание равновесия.
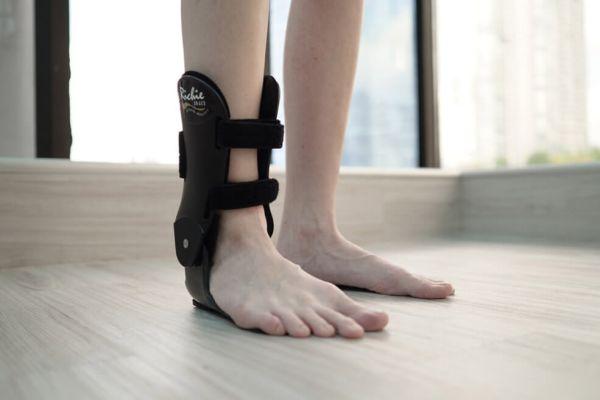
Использование ортезов ассоциируется с длительной двигательной активностью, предотвращением падений и травм, способностью к самообслуживанию. Ортезы поддерживают стопу в физиологическом положении. Пациенты, которые регулярно носят ортезы, самостоятельно передвигаются, не нуждаются в посторонней помощи.
Прогноз
Болезнь Шарко-Мари-Тута не сказывается на продолжительности жизни, пациенты живут столько, сколько позволяет общее состояние здоровья. Прогноз при невральной амиотрофии относительно благоприятный, корректная терапия улучшает двигательные функции пациента и препятствует развитию осложнений (утрата способности самостоятельно передвигаться, ухаживать за собой, принимать пищу, писать).
Пациентам рекомендуется избегать чрезмерных физических нагрузок, которые провоцируют усиление симптоматических проявлений. Полезны дозированные занятия видами спорта – ходьба, пилатес, плавание, езда на велосипеде. Необходимо организовать правильное питание, богатое витаминами, минералами и другими нутриентами.
Пациенту важно следить за показателями массы тела. Избыточный вес ассоциируется с дополнительными нагрузками на ослабленные мышцы, болезненными ощущениями в области ног и поясничного отдела позвоночного столба. Проводится диагностическое обследование членов семей пациентов с диагностированной БШМТ для выявления носителей генных мутаций для последующего назначения им своевременного лечения.
Заболевание Шарко-Мари-Тута относится к наследственным патологиям, поражающим периферические нервы. Невральная амиотрофия приводит к расстройству чувствительности в дистальных сегментах конечностей и нарушению двигательной активности.
Читайте также:
