
Семейная форма болезни альцгеймера
Болезнь Альцгеймера представляет собой одно из наиболее частых нейродегенеративных заболеваний человека и ведущую причину деменции в современном обществе [Rocca W. et al., 1991; Schellenberg G., 1995]. Ее распространенность у лиц старше 65 лет составляет около 3% и неуклонно повышается в более пожилых возрастных группах [Katzman R., 1976]. На секции выявляется диффузная атрофия мозга, с большей выраженостью изменений в теменных и затылочных областях.
При микроскопическом исследовании обнаруживаются два основных гистологических признака болезни Альцгеймера: 1) амилоидные (сенильные) бляшки в паренхиме мозга, главным компонентом которых являются фибриллярные агрегаты гидрофобного пептида (3-амилоида, состоящего из 39-43 аминокислот; 2) нейрофибриллярные клубки, представляющие собой спаянные попарно скрученные филаменты измененных нейронов, легко выявляемые при импрегнации серебром [McKee A. et al., 1991]. Характерно также выпадение нейронов, глиоз, наличие единичных телец Леви. Отмеченные изменения наиболее выражены в коре больших полушарий и гиппокампе.
Интересно отметить, что мутация Glu693Gln в 17-м экзоне гена АРР приводит к развитию другого заболевания с близким патогенезом - так называемого наследственного церебрального кровоизлияния голландского типа, обусловленного изменением стенок церебральных сосудов вследствие отложения в них амилоидных депозитов [Levy E. et al., 1990]. Более того, оба АРР-ассоциированных заболевания (деменция альцгеймеровского типа и церебральное кровоизлияние) могут наблюдаться у различных родственников в одной и той же родословной -как это описано в семье с мутацией Gly692Ala в 17-м экзоне гена АРР [Hendriks L. et al., 1992].
Принимая во внимание, что аутосомно-доминантные формы болезни Альцгеймера характеризуются чаще всего ранним началом заболевания, с практической точки зрения мутационный скрининг целесообразно ограничивать семьями, в которых хотя бы у части родственников симптомы болезни манифестировали до 60 лет. Правильному выбору гена, подлежащего исследованию, может способствовать предварительное установление сцепления болезни с одним из известных локусов на хромосомах lq, 14q или 21q. Если такой анализ сцепления невозможен ввиду небольшого размера обследуемых семей, мутационный анализ генов проводится в следующей последовательности: пресенилин-1, АРР, пресенилин-2 (в соответствии с частотой встречаемости мутаций в данных генах).
Поиск мутаций в пресенилипах проводится как на геномной ДНК (SSCP-анализ + прямое секвснирование отдельных экзонов), так и на ДНК из лимфоцитов крови, которая амплифицируется в виде полного набора перекрывающихся фрагментов и подвергается тотальному секвенированию. Для ДНК-диагностики мутаций в гене АРР обычно избирательно секвенируются 16-й и 17-й экзоны гена, кодирующие структуру основного патогеннного продукта белка АРР - пептида А(3. При исключении мутаций в исследованных областях всех указанных генов некоторые авторы рекомендуют также секвенировать экзоны 7, 8 и 18 гена АРР, соответствующие функционально значимым участкам АРР-белка.
По данным разных авторов, изучавших различные популяции и выборки больных с ранними аутосомно-доминантными случаями болезни Альцгеймера, общая частота выявления мутаций в генах АРР и пресенилинов варьирует от 18% до 100% [Hutton ML, Hardy J., 1997; Cruts M. et al., 1998; Campion D. et al., 1999]. Поскольку даже в семьях с подтвержденным сцеплением секвени-рование может не выявлять нарушений нуклеотидного состава кодирующей части гена (это возможно при локализации мутаций в интронах, промоторной области и др.), в таких семьях доступным методом молекулярной диагностики может служить косвенная ДНК-диагностика с использованием маркеров мутантного хромосомного локуса.

Этиология и патогенез болезнь Альцгеймера еще полностью не изучены. Научные исследования подтверждают генетическую природу этого заболевания в 70% случаев, в остальных 30% она остается неизвестной. Это означает, что могут существовать другие причины ― факторы внешней среды или же генные мутации, еще не обнаруженные учеными.
Анализ ДНК в рутинной диагностике Альцгеймера не применяют, это нецелесообразно с финансовой точки зрения и не имеет существенного значения для выбора тактики лечения. Генетические исследования необходимы ученым для поиска методов лечения. Иногда к таким анализам прибегают для выявления рисков развития заболевания.

Генетическое заболевание или генетическая предрасположенность?
Между этими понятиями имеется существенная разница. Когда болезнь называют генетической, имеют в виду, что при наличии определенного гена в структуре человеческой ДНК она обязательно проявится.
Наследственные виды этого заболевания развиваются из-за мутации в тех генах, которые отвечают за синтез белков в центральной нервной системе. Патогенез болезни Альцгеймера наследственного типа изучен лучше всего. В результате мутаций и последующих сбоев в работе белок-синтезирующих систем накапливается патологическое вещество ― бета-амилоид, бляшки из которого находят при посмертном вскрытии мозга больных.

Важно! На сегодняшний день ученые склоняются к тому, что большинство форм патологии появляются из-за генетической предрасположенности. То есть в их развитии участвует не только генетика, но и внешние факторы.
Поэтому шанс того, что у родственника больного Альцгеймер тоже проявится, невелик. В случае чисто генетической формы заболевания эта вероятность может достигать 50%.
Как может быть унаследован Альцгеймер?
По результатам многочисленных исследований членов семей больных и дальнейших наблюдений за испытуемыми было выявлено два основных вида наследования патологии: аутосомно-доминантная семейная форма и другие формы.
Причина заболевания ― редкий тип генной мутации, который передается по наследству из поколения в поколение. В таких семьях недуг обнаруживается в 50% случаев. Первые симптомы проявляются рано (до 55 лет).
Чтобы подтвердить семейную форму, прибегают к генетическому анализу ДНК больного и родственников, составляют генеалогическое древо с указанием всех известных случаев деменции в семье. При таких формах заболевания люди с данной патологией встречаются в каждом поколении.

В большинстве случаев она обусловлена не одной наследственной мутацией, а множественными изменениями в геноме, которые не наследуются. Этиология болезни Альцгеймера такого типа не изучена, а многие теории ставятся под сомнение.
Значительную роль в процессе развития болезни играют и внешние факторы, такие как экология, питание, влияние социума, условия труда.
Важно! В развитии Альцгеймера гены играют не самую важную роль, образ жизни человека, его физическая активность и питание во многом определяют скорость развития патологических процессов в центральной нервной системе.

При таких формах заболевания не отслеживается закономерность в генеалогическом древе. Если его проанализировать, то случаи деменции будут встречаться в виде единичных случаев и не в каждом поколении.
Тактика обследования родственников пациентов с Альцгеймером
Если у человека диагностирован Альцгеймер, то родственникам больного следует провести консультацию с лечащим врачом по вопросам ухода за ним, а также рисков развития недуга у других членов семьи. В ходе беседы с членами семьи врач должен выяснить анамнез, частоту развития деменции в семье.
Показания к консультации врача-генетика:
- 3 и более случая диагностированной деменции среди семьи;
- установление диагноза у кровных близких родственников (ребенок-родитель, братья-сестры);
- множественные случаи ранней смерти в семье больного;
- раннее начало болезни (диагностирована до 55 лет).
Во всех этих случаях можно заподозрить аутосомно-доминантную семейную форму Альцгеймера. Генетик должен опросить всех близких членов семьи, составить подробное генеалогическое древо, опираясь на данные из официальных медицинских документов, выписок из амбулаторных карт и психиатрических заключений. Такую информацию бывает довольно проблематично собрать, поэтому генеалогический анализ может давать погрешность.

Допускается возможность применения генетического анализа ДНК родственников больного. Для этого должен быть точно установлен ген, в котором произошла мутация. Затем в биологическом материале обследуемых родственников устанавливается наличие такого гена. При помощи такого теста можно с максимальной точностью (почти в 100% случаев) установить вероятность развития болезни. Однако это дорогостоящий метод, пока что он применяется только в рамках клинических исследований.
Если оснований подозревать семейную форму заболевания нет, то нельзя предсказать риски развития ее у родственников. Так, шансы выявления болезни возрастают с возрастом независимо от того, были ли подобные случаи уже в семье.
| Возраст | Заболеваемость среди данной возрастной группы |
|---|---|
| 65-69 | 1,5% |
| 70-74 | 3,5% |
| 75-79 | 6,8% |
| 80-84 | 13,6% |
| 85-89 | 22% |
| 90-94 | 32% |
| Старше 95 | 45% |
Так, отслеживается прямая взаимосвязь между возрастом и частотой возникновения деменции. То есть все люди имеют риск заболеть при условии, что они проживут достаточно долго. Эта закономерность не отслеживается в группах больных с семейными формами Альцгеймера, при которых первые симптомы патологии проявляются рано (от 20 до 50 лет).

Итак, к какой группе патологий можно отнести болезнь Альцгеймера, генетическое заболевание это или нет? Чисто генетическими можно назвать только семейные формы заболевания, которые встречаются довольно редко, они характеризуются ранним началом и более стремительным течением.
В подавляющем большинстве случаев эту болезнь нельзя назвать генетической. Это многофакторная патология, которая характерна для стареющего организма, кого-то она настигает раньше, кого-то позже, кто-то не доживает до момента установки диагноза.

Видео











Особенности семейной формы болезни Альцгеймера с ранним началом
Болезнь Альцгеймера (БА) – наиболее распространенная форма деменции в клинической практике. Хуже диагностируется ранняя форма БА в связи с редкой встречаемостью. В данной статье представлен актуальный обзор данных о частоте встречаемости, патоморфологии головного мозга, клинической картине, диагностике и медикаментозном лечении БА с ранним началом. Особое внимание уделено генетическим аспектам БА с аутосомно-доминантным типом наследования.
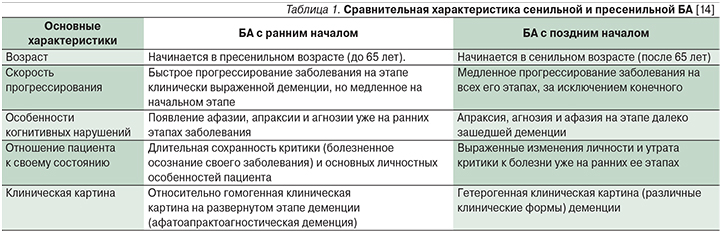
Болезнь Альцгеймера (БА) – нейродегенеративное заболевание, проявляющееся прогрессивным снижением памяти и других когнитивных функций вследствие гибели нейронов коры больших полушарий [1]. БА с ранним началом (до 65 лет) составляет 1–5% от всех случаев БА [2]. В США насчитывают примерно 200 тыс. пациентов с ранней формой БА, что как раз составляет около 4% от 5,7 млн больных БА [3]. В российских исследованиях данные о заболеваемости пациентов с БА с ранним началом отсутствуют. По данным Росстата, смертность от БА с ранним началом за 2012 г. у пациентов в возрасте 35–44, 45–54 и 55–64 года составила 0,005, 0,038 и 0,330 на 100 тыс. населения соответственно [4]. Актуальность проблемы заключается не только в том, что ранняя форма хуже диагностируется в связи с редкой встречаемостью, протекает более быстро и агрессивно, а также длительно осознается пациентом по сравнению с БА с поздним началом (табл. 1), но и в том, что пациентами становятся лица трудоспособного возраста, что способствует формированию разнообразных психологических, социальных и экономических проблем [5].
В настоящее время понятие БА с ранним началом расширено и дополнено. БА с ранним началом может иметь как семейную, так и спорадическую формы [6]. Причем около половины случаев семейной формы БА с ранним началом приходится на аутосомно-доминантную форму [7]. Семейная форма БА может относиться к заболеванию не только с ранним, но и с поздним началом (после 65 лет). Причем часть случаев семейной формы БА с поздним началом приходится также на аутосомно-доминантную форму [8]. Таким образом, существует несколько сходных, в определенной степени перекрывающихся состояний: семейная форма БА (FAD – Familial Alzheimer’s Disease) [9], БА с ранним началом (EOAD – Early-Onset Alzheimer’s Disease) [10], семейная форма БА с ранним началом (EOFAD – Early-onset familial Alzheimer’s disease) [11], аутосомно-доминантная БА (ADAD – autosomal dominant Alzheimer’s disease) [12] и аутосомно-доминантная форма БА с ранним началом (ADEOAD – early-onset autosomal dominant Alzheimer’s disease) [13].
В настоящей статье речь пойдет о семейной форме БА с ранним началом, включая аутосомно-доминантную форму.
БА с ранним началом обычно начинается в возрасте от 35 до 65 лет. Средний возраст больных составляет 54–56 лет, средняя продолжительность жизни после установления диагноза – 8–10 [15]. Заболевание сопровождается прогрессирующим нарушением памяти, интеллектуальной деятельности и высших корковых функций, приводит к развитию тотальной деменции с синдромами афазии, апраксии и агнозии [16]. Возможно проявление зрительной дисфункции и дискалькулии [17].
Также, по данным И.В. Колыхалова (2016), у 60% пациентов на том или ином этапе развития заболевания имеют место поведенческие и психопатологические расстройства следующих групп:
- Психотические расстройства (бредовые – 31%, зрительные и слуховые галлюцинации – 13 и 10% соответственно);
- Аффективные расстройства (депрессия – более 50%, тревога – от 24 до 65%, апатия – от 19 до 76%);
- Поведенческие нарушения (агрессия – 6%, двигательное беспокойство [аберрантное моторное поведение] – 12–84%) [18].
Патоморфологическая картина характеризуется церебральной атрофией с уменьшением объема и массы мозга, атрофией извилин коры, c расширением корковых борозд и желудочковой системы. При микроскопическом исследовании отмечается массивная утрата нейронов коры мозга, гиппокампа, а также базального ядра Мейнерта и голубого пятна ствола мозга. У сохранившихся нейронов выявляется дегенерация дендритов [19].
В связи с тем что семейная форма БА может дебютировать в разное время (быть БА с ранним или поздним началом), исследователи уделяют большое внимание этиологическим факторам развития заболевания. Считается, что чем позже наступает БА, тем больше доминируют факторы старения и окружающей среды (лентивирусы, дефекты энергетического метаболизма, недостаточность нейротрофических факторов, эксайтотоксичность, митохондриальные дефекты, нейротоксичность микроэлементов и окислительный стресс [20]) и тем меньше влияет генетическая предрасположенность. А чем раньше возраст начала, тем БА будет больше определяться детерминированной мутацией в одном гене [21].
БА с ранним началом – заболевание практически с полной генетической детерминированностью, наследуемость составляет от 92 до 100% [22].
У 35–60% пациентов с БА с ранним началом обнаруживается не менее одного родственника первой степени родства с данным заболеванием (т.е. семейная форма БА с ранним началом), причем у 10–15% пациентов с семейной БА с ранним началом отмечается аутосомно-доминантный тип наследования [23]. Аутосомно-доминантная форма БА с ранним началом составляет 0,5% от всех случаев БА [9].
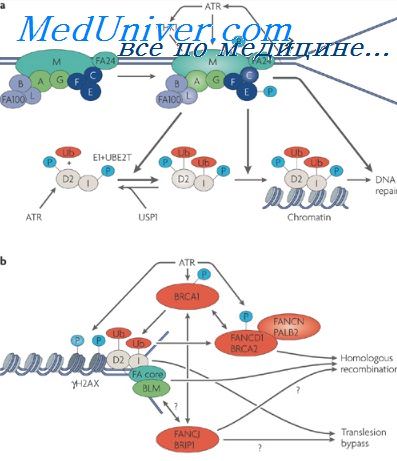
Почти у 50% родственников пациентов с симптомами аутосомно-доминантной формы БА с ранним началом выявляются мутации в следующих генах [24] (табл. 2):
- ген белка – предшественника амилоида (APP – amyloid precursor protein) локализован на 21-й хромосоме; тип наследования аутосомно-доминантный с полной пенетрантностью;
- ген белка пресенилина 1 (PSEN1 – presenilin protein 1) локализован на 14-й хромосоме; тип наследования аутосомно-доминантный с неполной пенетрантностью;
- ген белка пресенилина 2 (PSEN2 – presenilin protein 2), локализующегося на 1-й хромосоме; тип наследования аутосомно-доминантный с неполной пенетрантностью.
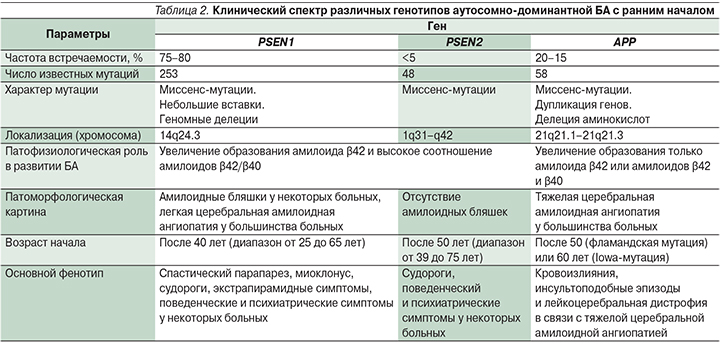
Мутации гена PSEN1 ответственны за 75–80% случаев развития БА с ранним началом, мутации генов APP и PSEN2 – за 20–15% и 0
Болезнь Альцгеймера — это хроническое прогрессирующее заболевание, которое ухудшает память, мышление и движение. Некоторые исследования показывают, что человек подвергается повышенному риску развития болезни Альцгеймера, если у его родственника есть это расстройство.
Болезнь Альцгеймера — самая распространенная причина деменции (слабоумия), которая может серьезно повлиять на способность человека думать, выносить суждения и выполнять повседневные задачи. Врачи знали о болезни Альцгеймера в течение многих лет, но многие аспекты этого состояния и возможное лечение остаются неизвестными. Причины болезни Альцгеймера неясны. Современные исследования показывают, что развитию болезни Альцгеймера может способствовать множество факторов, одним из которых является генетика или наследственность.
Передается ли болезнь Альцгеймера по наследству?
Ученые описывают генетические риски болезни Альцгеймера в терминах двух факторов: гены риска и детерминированные гены.
Когда у человека есть гены риска, это означает, что у него повышается вероятность развития болезни. Например, женщина с генами BRCA1 и BRCA2 имеет более высокий риск развития рака молочной железы.
Детерминированные гены могут непосредственно вызывать развитие болезни.
Ученые обнаружили несколько генов риска и детерминизма для болезни Альцгеймера.
Гены риска
Несколько генов представляют собой риск для развития болезни Альцгеймера. Ген с наиболее существенной связью с болезнью Альцгеймера — ген аполипопротеина E-е4 (APOE-e4). Примерно 20-25 % людей с этим геном могут страдать болезнью Альцгеймера. Человек, унаследовавший ген APOE-e4 от обоих родителей, имеет более высокий риск развития болезни Альцгеймера, чем человек, унаследовавший ген только от одного. Наличие гена может означать, что человек проявляет симптомы в раннем возрасте и ему ставят более ранний диагноз. Другие гены могут оказывать влияние на позднюю стадию болезни Альцгеймера и ее развитие. Ученым необходимо провести дополнительные исследования, чтобы узнать, как эти гены увеличивают риск развития болезни Альцгеймера.
Некоторые из этих генов регулируют определенные факторы в мозгу, такие как воспаление и способ общения нервных клеток. В то время как каждый человек наследует ген APOE в той или иной форме, гены APOE-е3 и APOE-е2 не имеют никакой связи с болезнью Альцгеймера. APOE-е2 может даже оказывать защитное воздействие на мозг от этой болезни.
Детерминированные гены
Исследователи выделили три специфических детерминированных гена , которые могут вызывать болезнь Альцгеймера:
- предшественник бета-амилоида (APP)
- пресенилин-1 (PS-1)
- пресенилин-2 (PS-2)
Эти гены ответственны за чрезмерное накопление амилоидного бета-пептида — токсичный белок, который скапливается в мозгу, и вызывает повреждение нервных клеток, что характерно для болезни Альцгеймера. Однако не все люди с ранним началом болезни Альцгеймера имеют эти гены. Человек с этими генами, у которого развивается болезнь Альцгеймера, имеет редкий тип, известный как семейное расстройство Альцгеймера. Семейная болезнь Альцгеймера составляет менее 5% всех случаев в мире. Болезнь Альцгеймера, вызванная детерминированными генами, обычно возникает в возрасте до 65 лет. Иногда она может развиться у людей в возрасте 40-50 лет.
Влияние генов на другие виды деменции
Некоторые виды деменции связаны с другими генетическими пороками развития.
Болезнь Хантингтона , например, вызвана изменением 4 хромосомы, что может проявляться прогрессирующим слабоумием. Болезнь Хантингтона является доминирующим генетическим заболеванием. Это означает, что если у одного из родителей есть заболевание, они могут передать ген своему потомству, и у них разовьется болезнь. Симптомы болезни Хантингтона обычно не проявляются до 30-50-летнего возраста. Это может затруднить врачам прогнозирование и диагностику до того, как у человека появятся дети и он передаст ген.
Исследователи полагают, что деменция с тельцами Леви или деменция при болезни Паркинсона , также может иметь генетический компонент. Однако они полагают, что другие факторы, не связанные с генетикой, также могут играть определенную роль в развитии этих состояний.
Факторы риска развития болезни Альцгеймера
Исследователи выявили несколько факторов риска развития болезни Альцгеймера. К ним относятся:
Возраст : наиболее значимым фактором риска развития болезни Альцгеймера является возраст. У людей старше 65 лет вероятность развития болезни Альцгеймера выше, чем у молодых людей. К 85 годам 1 из 3 человек страдает этим заболеванием.
Семейный анамнез : наличие близкого родственника с болезнью Альцгеймера увеличивает риск ее развития.
Травма головы : люди с предыдущими случаями тяжелой травмы головы, например, в результате автомобильной аварии или контактных видов спорта, подвержены более высокому риску развития болезни Альцгеймера.
Здоровье сердца : проблемы со здоровьем сердечно-сосудистой системы могут увеличить вероятность развития болезни Альцгеймера. Примеры включают высокое артериальное давление , ишемический инсульт , диабет, болезни сердца и высокий уровень холестерина . Перечисленные выше состояния могут повредить кровеносные сосуды в головном мозгу, что влияет на риск развития болезни Альцгеймера.
Ранние признаки болезни Альцгеймера
Болезнь Альцгеймера обычно сопровождается постепенной потерей памяти и функций мозга. Ранние симптомы могут включать периоды забывчивости или потери памяти. Со временем человек может испытывать замешательство или дезориентацию в привычных условиях, в том числе и дома.
Другие симптомы могут включать:
- изменения в настроении или личности
- путаница во времени и месте
- трудности с рутинными задачами, такими как стирка или приготовление пищи
- трудность распознавания объектов
- трудности с распознаванием людей
Процесс старения может естественным образом ухудшить память человека, но болезнь Альцгеймера приводит к более длительным периодам забывчивости.
Со временем человеку с болезнью Альцгеймера может потребоваться все больше помощи в повседневной жизни, такой как чистка зубов, одевание и прием пищи. Они могут испытывать волнение, беспокойство, личностные расстройства и проблемы с речью. Продолжительность жизни человека с болезнью Альцгеймера обычно составляет от 8 до 10 лет после появления первых симптомов заболевания.
Поскольку люди с прогрессирующей болезнью Альцгеймера не могут заботиться о себе или могут не осознавать важность еды, распространенные причины смерти включают недоедание, истощение организма или пневмонию.
Когда нужно обратиться к врачу
Обращение за медицинской помощью для человека, который имеет эти симптомы, имеет жизненно важное значение. Врач может исключить другие состояния, которые могут вызвать деменцию, такие как инфекция мочевыводящих путей или опухоль головного мозга.
Члены семьи должны составить список всех лекарств, которые в настоящее время принимает человек с симптомами заболевания. Врач может просмотреть список и убедиться, что симптомы деменции не являются побочными эффектами лекарственных средств.
Определение заметных симптомов и их развития может помочь врачу установить потенциальные закономерности. Хотя генетическое тестирование доступно, врачи обычно его не рекомендуют. Наличие генов не обязательно означает, что у человека будет такое состояние, а тестирование может вызвать ненужное беспокойство и страх. Однако человек с семейным анамнезом ранней стадии болезни Альцгеймера может пожелать пройти генетическое тестирование. Большинство врачей рекомендуют заранее встретиться с генетическим консультантом, чтобы обсудить плюсы и минусы генетического тестирования и то, как они могут интерпретировать результаты.
Иногда врач может рекомендовать генетическое тестирование, когда у человека проявляются ранние симптомы болезни Альцгеймера, поскольку это может диктовать возможные методы лечения.
Заключение
Болезнь Альцгеймера связана с рядом генов. Некоторые из них, такие как ген APOE-e4, повышают риск развития заболевания, но не всегда приводят к болезни Альцгеймера. Ген APP непосредственно вызывает развитие болезни. Однако это редкий тип, известный как семейная болезнь Альцгеймера, которая встречается менее чем у 5% людей с этим заболеванием. В настоящее время ученые проводят несколько крупномасштабных исследований болезни Альцгеймера и ее связи с наследственностью.

Приглашаем подписаться на наш канал в Яндекс Дзен
Синдром Альцгеймера, или болезнь Альцгеймера (БА), — очень распространенная патология. Как известно, это дегенеративное заболевание головного мозга с прогрессирующим слабоумием, которое развивается у пожилых людей.
Синдромом Альцгеймера болеют все — и обыкновенные люди, и всемирно известные. Например, бывший президент США Рональд Рейган, ирландская писательница Айрис Мёрдок, бывшая премьер-министр Великобритании Маргарет Тэтчер, актёр Питер Фальк, английский писатель Терри Пратчетт и ряд других знаменитостей имели или имеют сейчас диагноз болезни Альцгеймера.
Этиология заболевания до сих пор остается неясной, что особенно характерно в отношении ненаследственной (спорадической) формы БА. На фоне естественных процессов старения разворачиваются специфические для синдрома Альцгеймера события, центральные из которых — отложение -амилоида в коре и гиппокампе (Selkoe, 1994) и созревание сенильных бляшек. Это вызывает целый каскад молекулярных изменений, которые приводят к дисфункции и смерти нервных клеток по типу апоптоза или некроза, что клинически выражается прогрессирующей деменцией. В тканях мозга имеют место холинэргический дефицит, окислительный стресс, повышенный уровень провоспалительных цитокинов и глутамата (возбуждающего нейромедиатора).
Обнаружение причинных мутаций в генах белка-предшественника амилоида и пресенилина (b-АРР, PSEN-1 и) касаются очень редко встречающихся семей с аутосомно-доминантными формами заболевания, так называемой родословной семейной болезни Альцгеймера, которая поражает 50 % каждого поколения, независимо от пола, между 30 и 50 годами. Мутации в гене PSEN-2 характеризуются неполной пенетрантностью (т. е. не всегда приводят к развитию БА), они виновны в развитии более редких — как ранних, так и поздних — семейных форм болезни.
Изучение родословных несемейных форм БА дает такой результат: в среднем, риск развития БА для родственников первой степени родства составляет 3–14 %. При этом исследования близнецов показали значительную (но не 100 %) роль генетических факторов. Коэффициент конкордантности у монозиготных близнецов (85 %) значительно выше, чем у дизиготных (42 %).
Выделяют 2 формы БА — пресинильную (с началом до 65 лет), и сенильную (с началом после 65 лет). Крайне редко БА может дебютировать и в 30 лет (обычно это связано с мутацией гена пресенилина-1). Болезнь развивается некоторое время бессимптомно — мозгу удается компенсировать выпадение поврежденных нейронов. Клиника появляется, возможно, через годы после запуска патогенетического механизма. Ранним, хотя и недостаточно специфическим признаком болезни Альцгеймера, может служить выявляемая при магнитно-резонансной томографии атрофия медиальных отделов височных долей, прежде всего, гиппокампа. В диагностике БА может помочь нейропсихологическое скрининг-тестирование, при котором пациенты копируют фигуры, запоминают слова, читают, выполняют арифметические действия. Дифференциальную диагностику в первую очередь проводят с сосудистой деменцией.
К факторам риска БА в настоящее время относят:
- возраст
- женский пол (у женщин заболевание отмечается в два раза чаще, чем у мужчин)
- в анамнезе черепно-мозговая травма, инфаркт миокарда, гипотиреоз, депрессия, воздействие электромагнитных полей
- низкий уровень витамина В12 и фолатов в сыворотке крови
- курение
- отравление тяжелыми металлами
- длительный прием НПВС
- заместительная гормональная терапия (ЗГТ)
- возраст матери к моменту рождения индивидуума
- полный отказ от алкоголя или злоупотребление им
Защитными считаются следующие факторы:
- интеллектуальная работа, высокий уровень образования
- ежедневное употребление чая и кофе
- умеренное употребление алкоголя
Продолжительность жизни пациентов с момента постановки диагноза в среднем составляет 6–8 лет, но может варьироваться от 2 до 20 лет. Менее 3 % пациентов остаются в живых более четырнадцати лет. Первым симптом болезни Альцгеймера становится нарушение памяти. Сначала страдает запоминание нового материала, а профессиональная память нарушается в последнюю очередь. Снижается работоспособность, сужается круг интересов, появляется выраженная эмоциональная лабильность, тревожность, мнительность и некоторая расторможенность.
Позже присоединяются нарушения речи. Страдает в первую очередь понимание обращенной речи, способность же повторять слова и предложения сохраняется длительно. Кроме того, пациенты затрудняются называть предметы. Постепенно теряется навык осмысленного речевого общения, могут возникнуть эхолалии, палилалия или мутизм. Прогрессивно нарушаются праксис, гнозис и другие познавательных функции, изменяется личность, могут развиться аффективные расстройства и психозы (галлюцинации, бред).
Эхолалия — неконтролируемое автоматическое повторение слов, услышанных в чужой речи.
Паллилалия — болезненная потребность человека повторять некоторые слова или предложения.
Мутизм — отказ от речевого общения при отсутствии органических поражений речевого аппарата.
В финальной стадии больные утрачивают способность к самообслуживанию, дезориентированы в пространстве и времени — по сути, тотально слабоумны. Нередки неврологические симптомы: нарушение ходьбы, ригидность, брадикинезия, миоклонии, судорожные припадки, спастическая параплегия со сгибательной контрактурой ног. На терминальной стадии вследствие гипокинезии у пациентов возможно развитие сепсиса, урологических воспалительных заболеваний, аспирационной пневмонии, что может служить непосредственной причиной летального исхода. Неблагоприятные прогностические факторы, которые могут указывать на быстрое развитие болезни: экстрапирамидные нарушения (ригидность, брадикинезия, гипомимия, тремор покоя, а также нарушения ходьбы — шаркающая походка, ахейрокинезия, трудности поворотов).
У пациентов с пресенильным типом заболевания длительно сохраняются основные личностных особенностей, присутствует чувство собственной неполноценности или измененности и адекватное эмоциональное реагирование на болезнь. Типично медленное развитие болезни на инициальном и быстрое прогрессирование на этапе клинически выраженной деменции. Также для них характерно наличие богатой неврологической симптоматики.
В независимости от типа БА, в течение выделяют три стадии:
На стадии мягкой деменции пациент
Примерно у четверти пациентов на этой стадии имеются бредовые расстройства в виде эпизодических бредовых идей ущерба, воровства, реже — идей отношения, преследования или ревности.
На стадии умеренной деменции пациент
На стадии тяжелой деменции пациент
Увы, сегодня не существует лечения, которое могло бы предотвратить или существенно замедлить прогрессирование БА. Вот основные направления терапии и препараты.
2. Нейропротективная терапия помогает сохранять нейроны жизнеспособными, и должна применяться на начальных этапах болезни. Используют: а) антиоксиданты (витамин Е, экстракт гинкго билоба) — эффективнее на додементной стадии б) препараты с нейротрофическими свойствами (церебролизин) в) нейропротекторы (ницерголин)
Использование ноотропов (пирацетама, пиридитола) — препаратов, улучшающих церебральный метаболизм — не дало достоверных позитивных результатов при лечении больных БА. Применение больших доз нередко приводит к быстрому прогрессированию деменции (после кратковременного улучшения когнитивных функций). Этот эффект объясняется нейротрансмиттерным истощением холинергических структур после их чрезмерной стимуляции большими дозами ноотропов.
Гаврилова С. И., Жариков Г. А. Лечение болезни Альцгеймера // Психиатрия и психофармакотерапия Том 3/N 2/2001
4. Новые веяния в терапии БА Сегодня для лечения синдрома Альцгеймера начинают применять НПВС. Небольшое клиническое испытание индометацина показало, что у больных БА, получавших его в течение 6 месяцев, отмечалась стабилизация состояния, тогда как в группе больных, получавших плацебо, за этот период отмечено ухудшение по ряду параметров. Кроме того, опубликованные в 2004 году результаты клинических испытаний Национального института здоровья (National Institutes of Health (NIH)) селективных ингибиторов ЦОГ-2 (рофекоксиб, целекоксиб) показали, что они также обладают протективным действием и в то же время дают меньше нежелательных явлений, чем неселективные. Пока невозможно сказать, будут ли селективные НПВС столь же эффективны при БА, как неселективные.
Заместительная гормональная терапия — это доказанный фактор защиты от развития БА, однако она может иметь ряд серьезных осложнений (тромботические и онкологические). Сейчас только проверяется безопасность и эффективность ЗГТ при БА В стадии разработки находятся антиамилоидные стратегии терапии. Они развиваются по двум основным направлениям. Первое из них нацелено на нейтрализацию нейротоксических свойств b-амилоидных отложений посредством введения специальных лигандов или протеаз. Второе — на снижение продукции b-амилоида за счет ингибиторов ферментов синтеза амилоида.
Нашли ошибку? Выделите текст и нажмите Ctrl+Enter.
Читайте также:
