
Среди спиноцеребеллярных атаксий болезнь фридрейха отличается наличием
а) Атаксия Фридрейха. Атаксия Фридрейха — наиболее четко описанная и часто встречающаяся спиноцеребеллярная дегенерация. Частота встречаемости гена составляет 1:110 человек в Англии (Harding 1981a), и примерно один из 10000 человек в Швеции имеет клинические проявления.
Ген атаксии Фридрейха включает повторы ГА А последовательности в интроне 1, который распространен у пациентов (120-1700 повторов). Продуктом нормального гена является белок фратаксин, функция которого не полностью ясна. 94% пациентов с типичной атаксией Фридрейха являются гомозиготами по ГАА экспансии, тем не менее продолжительность повтора на каждой хромосоме из пары неодинакова (Durr et al., 1996a).
В редких случаях отмечается только одна мутация, но в такой ситуации выявляется точечная мутация в гомозиготном локусе (Campuzano et al., 1996). Выраженная длина повтора коррелирует с началом в раннем возрасте, более стремительным течением и наличием кардиомиопатии (Durr et al., 1996a).
Второй ген на хромосоме 9p23-p11 является причиной редких случаев (Фридрейха 2), клинически нечетко отличаемых от 1 типа (Christodoulou et al., 2001).
Основным патологическим проявлением является дистальная аксональная нейропатия, которая поражает нейроны длинных восходящих и нисходящих трактов спинного мозга и крупные сенсорные волокна периферических нервов и ганглии задних корешков (Said et al., 1986). Также зарегистрирована утрата нервных волокон в зрительных путях, а мозжечок остается непораженным.
Сердце увеличено, и более чем в половине случаев отмечается гипертрофическая кардиомиопатия с некрозом волокон и фиброзом, преимущественно затрагивающим левый желудочек.
Критерии диагностики атаксии Фридрейха (Harding, 1981a) включают начало до 25 лет (обычно до 16 лет), аутосомно-рецессивное наследование и сочетанное поражение крупных сенсорных волокон периферических нервов, мозжечкового тракта, пирамидного тракта и задних столбов.
Тем не менее, степень фенотипической вариабельности велика, в некоторых случаях отмечается позднее начало и/или меньшая выраженность симптомов и вариабельное течение, и некоторые пациенты прикованы к инвалидной коляске в раннем подростковом возрасте, в то время как другие способны самостоятельно передвигаться почти до 40 лет (Montermini et al., 1997).
По неофициальным данным, к доминантным случаям относится большая часть наследственной моторной и сенсорной нейропатии со скелетными деформациями и утратой чувствительности, но некоторые случаи не поддаются классификации.

Клинические проявления атаксии Фридрейха были описаны у 115 пациентов из 90 семей (Harding, 1981b). Основные проявления представлены в таблице ниже.
Заболевание чаще всего начинается в возрасте 5-16 лет, в редких случаях — в возрасте 2-5 лет. Прогрессирующая атаксия нижних конечностей с нарушением походки является основным проявлением, в то время как поражение верхних конечностей, приводящее к неуклюжести, в ранние сроки отмечается только в 25% случаев.
Сколиоз, тремор и изменения со стороны сердца редко являются первыми проявлениями, но формируются со временем, особенно при раннем начале заболевания. Конская стопа также является ранним симптомом. При осмотре в 70-95% случаев выявляется отсутствие глубоких рефлексов.
Дизартрия, пирамидные знаки со стороны ног и утрата глубокой и вибрационной чувствительности могут появляться позже, но относятся к постоянным симптомам. Нистагм встречается нечасто (20% случаев), медленные изломанные следящие движения глаз выявляются в 12% случаев (Harding, 1981b). Нестабильность фиксации является типичным проявлением (Alper и Narayanan, 2003).
Нередко встречается атрофия зрительного нерва, а глухота отмечается только у 10% пациентов. Дистальная атрофия отмечается практически в половине случаев. Интеллект не страдает.
Поражение сердца по результатам ЭКГ обнаруживается в трети случаев и даже чаще, если ЭКГ проводится систематически. Изменения зубца Т и аномалии сегмента ST являются ранними признаками сердечной недостаточности и единственным ее проявлением.
В конечном счете формируется прогрессирующая сердечная недостаточность или аритмия с фибрилляцией предсердий, половина пациентов умирает от сердечной недостаточности (Leone et al., 1988).
Течение заболевания медленное, но прогрессирующее. В среднем пациенты утрачивали способность ходить к 25 годам, со средней продолжительностью заболевания 15,5 лет.
Сахарный диабет является дальнейшим осложнением и развивается у 10% пациентов. Он имеет тенденцию сочетаться с атрофией зрительного нерва, в некоторых случаях диабетическая кома является причиной смерти.
Атипичные формы включают легкие случаи, которые, возможно, связаны с одним и тем же локусом 9-й хромосомы. К данной группе относятся случаи сохранения сухожильных рефлексов (Palau et al., 1995) и поздние формы с началом в раннем взрослом возрасте (De Michele et al., 1994).
Результаты одной из недавних работ, в которой использовалось возможное выявление мутантного гена, предполагают, что клиническая картина более вариабельна, чем считалось раньше (Palau et al., 1995; Pandolfo 2003). 25% пациентов в рамкам одного крупного исследования имели одно или более атипичное проявление (начало после 25 лет, сохранение или даже оживление сухожильных рефлексов или отсутствие симптома Бабинского).
Возраст начала превышал 20 лет у 19 из 114 пациентов (De Michele et al., 1994). Сохраненные рефлексы среди пациентов, которые в остальном соответствуют всем критериям, зарегистрированы у значимого числа пациентов (Palau et al., 1995). Большая часть случаев, ранее отнесенных к рано начинающейся атаксии с сохранением глубоких сухожильных рефлексов (состояния, отличного от атаксии Фридрейха) (Harding, 1981b; Klockgether et al., 1991), по результатам молекулярно-генетических исследований, вероятно, имеют отношение к болезни Фридрейха.
Эта группа, очевидно, была гетерогенной как при раннем ( 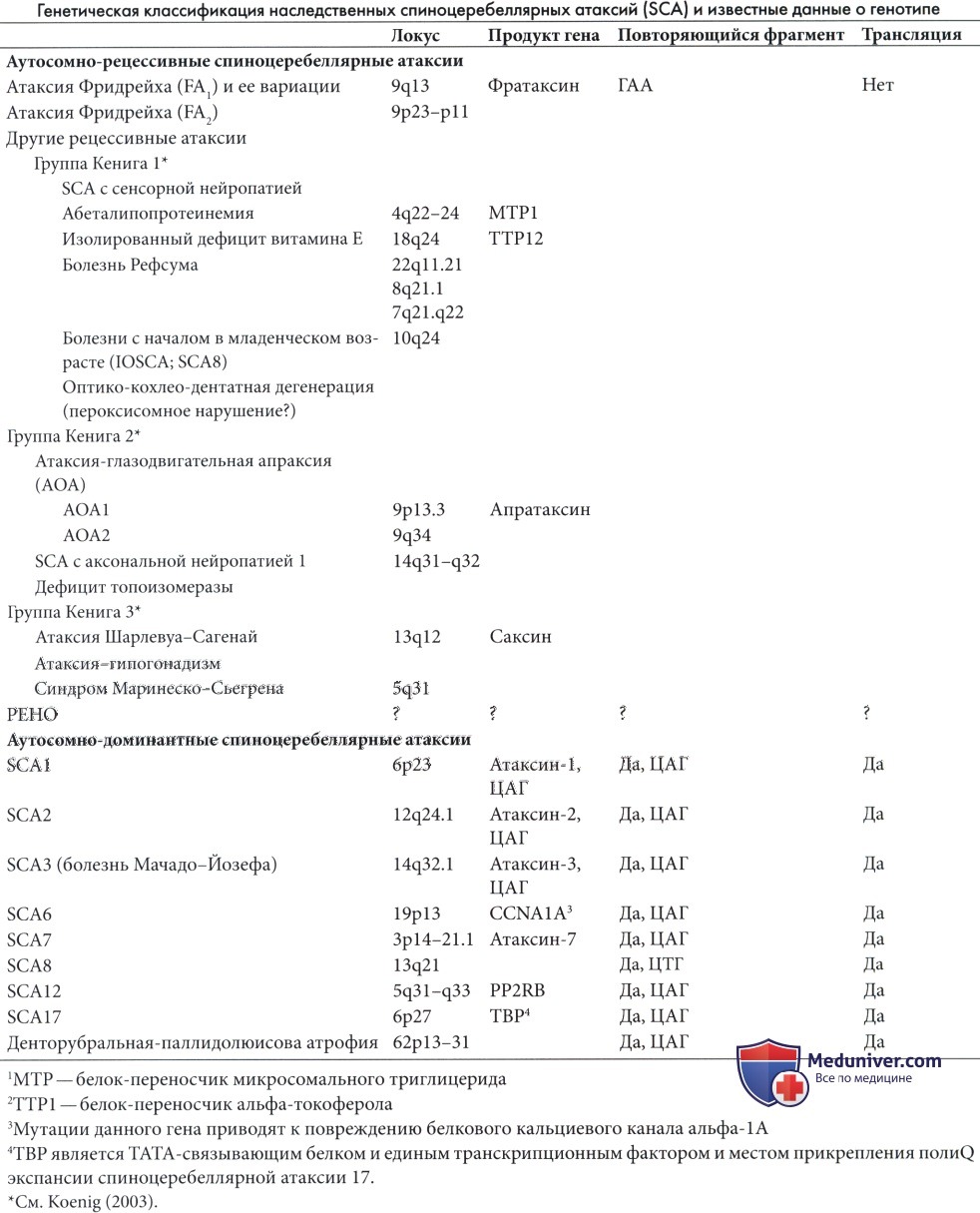
Вторая подгруппа Кенига включает атаксию со зрительной моторной апраксией (АОА), которая делится на два типа: AOA1 является одной из наиболее распространенных форм детского возраста и описана вместе с атаксией-телеангиэктазией, несмотря на то, что ее физиология кажется более сходной с спиноцеребеллярными атаксиями (SCA, см. далее). Одним из важных биологических признаков является гипоальбуминемия, которая практически постоянно обнаруживается и имеет диагностическую значимость. АОА 2 типа встречается реже и начинается позже (в позднем подростковом или раннем взрослом возрасте). Умеренно повышенный уровень альфа-фетопротеина отмечается в 75% случаев.
Клинические проявления AOA1 очень напоминают проявления атаксии-телеангиэктазии, но без признаков экстраневрологических поражений.
В отличии от атаксии-телеангиэктазии, AOA1 не связана с повышением уровня альфа-фетопротеина, хромосомными аномалиями, склонностью к раковым опухолям или повышенной радиочувствительностью культуры фибробластов (Le Ber et al., 2005). Редким, но интересным состоянием является спиноцеребеллярная атаксия с аксональной нейропатией 1 (SCA1), которая фактически является нарушением репарации ДНК, вызванной отсутствием фермента топоизомеразы-фосфодиэстеразы-1 (TDP1) (E1-Khamisy et al., 2005).
В третьей подгруппе Кенига четко описана атаксия Шарлевуа-Сагеней. Изначально синдром был описан в Квебеке, но с тех пор регистрировался и в других частях света (Gucuyener et al., 2001). Заболевание связано с мутацией гена сакцина на 13-й хромосоме (Engert et al., 2000). Фенотипические проявления включают заметную спастичность и постоянное наличие полос на глазном дне с преобладанием миелиновых волокон, радиально расходящихся от диска зрительного нерва.
Два редких аутосомно-рецессивных синдрома включают очень медленно прогрессирующую атаксию и могут рассматриваться вместе с SCA. Несмотря на то, что патология и механизмы заболеваний отличаются, они проявляются несколькими общими клиническими симптомами.
Синдром Маринеску-Шегрена включает атрофию мозжечка, преимущественно затрагивающую червь, раннее начало медленно прогрессирующей атаксии, катаракту, легкую задержку умственного развития, иногда гипогонадизм (Sewry et al., 1988) и позднее развитие специфической миопатии (Superneau et al., 1987). Заболевание развивается в результате мутации гена SLI1, кодирующего белок-шаперон, ключевой регулятор основных функций эндоплазматической сети.
РЕНО синдром (прогрессирующая энцефалопатия с периферическими отеками, гипсаритмией и атрофией зрительного нерва, также называемая церебелло-оптический синдром) является рецессивным заболеванием, зарегистрированным преимущественно в Финляндии (Salonen et al., 1991), хотя регистрировались случаи и в других странах. Основными проявлениями являются рано начинающиеся припадки, легкий дисморфизм, периферические отеки и регрессия, начинающаяся в возрасте 3-5 месяцев. Атрофия зрительного нерва развивается к концу первого года (Haltia и Somer, 1993).
- Вернуться в оглавление раздела "Неврология."
Редактор: Искандер Милевски. Дата публикации: 18.12.2018
001. Основными задачами медицинской генетики является изучение
- законов наследственности и изменчивости человеческого организма
- популяционной статистики наследственных заболеваний
- молекулярных и биохимических аспектов наследственности
- ничего из перечисленного
002. Доминантный признак по закону Менделя проявляется при скрещивании во втором поколении с частотой
003. Доминантный ген-это ген, действие которого
- выявляется в гетерозиготном состоянии
- выявляется в гомозиготном состоянии
+ выявляется в гетеро- и гомозиготном состоянии
- неверно все из перечисленного
- верно все из перечисленного
004. Генотип организма представляет собой систему взаимодействия генов, при которой наследственные признаки определяются путем участия
- одного гена в определении одного признака
- одного гена в определении многих признаков
- многих генов в определении одного признака
+ верно все перечисленное
- ничего из перечисленного неверно
005. Пробандом называют
- здорового носителя мутантного гена
+ больного носителя мутантного гена
- здорового родителя больного с признаками наследственного заболевания
- ребенка, больного наследственным заболеванием
- нет верного ответа
006. Сибсом называют
- здорового родителя больного наследственным заболеванием
- ребенка больного наследственным заболеванием
+ родного брата или сестру (но не близнецов) больного наследственным заболеванием
- любых родственников больного
007. Фенотип-это совокупность признаков и свойств организма, проявление которых обусловлено
- действием доминантного гена
- действием рецессивного гена
+ взаимодействием генотипа с факторами среды
- взаимодействие половых хромосом
008. Кариотип-это совокупность особенностей хромосомного набора (комплекс- клетки, определяющаяся
- числом половых хромосом
- ничем из перечисленнного
009. Аутосомно-доминантный тип наследования отличается
- преимущественным поражением лиц мужского пола
- преобладанием в поколении больных членов семьи
+ проявлением патологического наследуемого признака во всех поколениях без пропуска
- преимущественное поражение лиц женского пола
- проявлением наследственного признака не во всех поколениях
010. Аутосомно-рецессивный тип наследования отличается тем, что
- соотношение здоровых и больных членов семьи равно 1:1
- заболевание не связано с кровным родством
+ родители первого выявленного больного клинически здоровы
- один из родителей больного болен
- родители перкого выявленного больного больны
011. Рецессивный тип наследования, связанный с Х-хромосомой (сцепленный с полом), отличается тем, что
- соотношение больных мужчин в каждом поколении равно 2:1
+ заболевают только мужчины
- заболевают только женщины
- признаки болезни обязательно находят у матери пробанда
- заболевают и мужчины и женщины
012. Причиной хромосомных заболеваний могут быть
- изменения числа хромосом
- изменения размера хромосом
- нарушения структуры хромосом
- ничего из перечисленного
013. Фенотипическими признаками хромосомных болезней являются
- нарушения психического развития
- нарушения физического развития
- множественные пороки развития
- ничего из перечисленного
014. Индуцированный мутагенез вызывают следующие факторы
- хронические интоксикации тяжелыми металлами
+ все перечисленные факторы
015. В основу классификации наследственных болезней, учитывающей их генетическую природу, положены особенности
- количественных изменений хромосом
- ничего из перечисленного
016. Основным биохимическим признаком фенилкетонурии является повышение содержания
017. При поздней форме амавротической идиотии Куфса у взрослых наблюдают
- ничего из перечисленного
018. Для болезни Дауна характерно сочетание следующих признаков
+ округлый череп, готическое небо, синдактилия, гипотония мышц
- долихоцефалия, расщепление неба, арахнодактилия, гипертонус мышц
- краниостенотический череп, заячья губа, наличие 6-го пальца, хореоатетоз
- наблюдается сочетание любых перечисленных признаков
- ничего из вышеперечисленного
019. Поражение нервной системы при лейкодистрофии происходит в результате
- избыточного накопления липидов в нервных клетках
- утраты липидов нервными клетками
+ распада липидов миелина и накопления продуктов распада в центральной нервной системе
- избыточного накопления холестерина в белом веществе
020. Для порфирии является характерным наличие
- порфобилиногена в моче
- повышенным содержанием порфирина в крови
021. Прогрессирующие мышечные дистрофии обусловлены поражением
- цереброспинальных пирамидных путей
- мотонейронов передних рогов спинного мозга
- задних корешков спинного мозга
022. Спинальная амиотрофия Верднига-Гоффмана наследуется
- по аутосомно-доминантному типу
+ по аутосомно-рецессивному типу
- по рецессивному типу, связанному с полом (Х-хромосома)
- по доминантному типу, связанному с полом
- сцепленное с Y-хромосомой, или голандрическое, наследование
023. Изменение контура ног по типу "опрокинутой бутылки" обусловлено изменением массы мышц
+ при амиотрофии Шарко-Мари-Тута
- при гипертрофической невропатии Дежерина-Сотта
- при мышечной дистрофии Эрба
- при мышечной дистрофии Беккера-Киннера
- при миотонической дистрофии
024. Амиотрофия Шарко-Мари-Тута обусловлена первичным поражением
- передних рогов спинного мозга
+ периферических двигательных нервов
- мышц дистальных отделов конечностей
- передних столбов спинного мозга
025. Тип наследования при амиотрофии Шарко-Мари-Тута характеризуется как
- сцепленный с полом (через Х-хромосому)
- ничего из вышеперчисленного
026. Прогрессирующая мышечная дистрофия формы Ландузи-Дежерина наследуется
+ по аутосомно-доминантному типу
- по аутосомно-рецессивному типу
- по рецессивному типу, сцепленному с полом (через Х-хромосому)
- ни по одному из вышеперечисленных типов наследования
- по всему перечисленному
027. Псевдогипертрофии наблюдают при следующих формах прогрессирующей мышечной дистрофии
- конечностно-поясная форма мышечной дистрофии
028. Тип наследования при миопатии Томсена характеризуется как
- Y-сцепленный с полом
029. При атрофической миотонии преобладает слабость мышц
- проксимальных отделов конечностей
- дистальных отделов конечностей
- мышц лица и проксимальных отделов конечностей
+ мышцы лица и дистальных отделов конечностей
030. Тип наследования при атрофической миотонии Штейнерта-Баттена характеризуется как
- сцепленный с полом (через Х-хромосому)
- сцепленный с полом (через Y-хромосому)
031. Тип наследования при гиперкалиемическом периодическом параличе характеризуется как
- сцепленный с полом (через Х-хромосому)
- сцепленный с полом (через Y-хромососу)
032. Тип наследования при гипокалиемическом периодическом параличе характеризуется как
- сцепленный с полом (через Х-хромосому)
- сцепленный с полом (через Y-хромососу)
+ доминантно и рецессивно
033. Нарушения медно-белкового обмена при гепатоцеребральной дистрофии Вильсона-Коновалова обусловлены дефектом гена следующей хромосомы
034. Для синдрома Шегрена-Ларссена не характерны
+ недостаточность слезо- и слюноотделения
- ничего из вышеперечисленного
035. При дрожательной и дрожательно-ригидной форме гепатоцеребральной дистрофии Вильсона-Коновалова преобладает тремор
- интенционный в руках
+ хлопающий в руках, статодинамический в туловище
036. Тип наследования при гепатоцеребральной дистрофии характеризуется как
- рецессивный, сцепленный с полом (через Х-хромосому)
- рецессивный, сцепленный с полом (через Y-хромосоу)
037. Приступ пароксизмальной миоплегии при гипокалиемической форме болезни Вестфаля-Шахновича обычно возникает
- во время тяжелой физической нагрузки
- сразу после тяжелой физической нагрузки
- в состоянии полного покоя днем
- во время ночного сна
038. Приступ миоплегии при гиперкалиемической (болезнь Гармсторп- и нормокалиемической форме (болезнь Посканцера и Керр- возникает
+ во время тяжелой физической нагрузки
- во время отдыха после физической нагрузки
- в состоянии покоя днем
- во время ночного сна
039. Тип наследования при хорее Гентингтона характеризуется как
- рецессивный, сцепленный с полом (через Х-хромосому)
- рецессивный, сцепленный с полом (через Y-хромосому)
040. Клиническая картина типичной хореи Гентингтона, кроме хореического гиперкинеза, включает
- пластическую экстрапирамидную ригидность
- симптом "зубчатого колеса"
041. Нейрохимические изменения в подкорковых ядрах при болезни Паркинсона характеризуются следующими изменениями моноаминов мозга
- увеличением содержания ацетилхолина
- увеличением содержанием ГАМК
- снижением содержания норадреналина
042. Болезнь Паркинсона может проявиться следующими синдромами
043. Тип наследования при синдроме Шегрена-Ларссена характеризуется как
- рецессивный, сцепленный с полом (через Х-хромосому)
- рецессивный, сцепленный с полом (Y-хромосому)
044. При наследственном эссенциальном дрожании тремор обычно имеет следующий характер
- все перечисленные варианты
045. Достаточными клиническими признаками в диагностике сирингомиелии являются
+ сегментарные диссоциированные нарушения чувствительности
- нижний спастический парапарез
- прогрессирующая атрофия мышц в участках, соответствующих сегментарным нарушениям чувствительности
046. При лечении болезни Паркинсона дофасодержащими средствами неврологические побочные симптомы проявляются
047. При лечении болезни Паркинсона холинолитиками (циклодол, норакин) побочные симптомы проявляются
+ затуманиванием зрения и сухость во рту
- двоением в глазах
048. Для переднероговой формы сирингомиелии характерны
- нарушение проприоцептивной чувствительности
+ ничего из перечисленного
049. Лечение холинолитиками болезни Паркинсона противопоказано при заболевании
050. Синдром Клиппеля-Фейля характеризуется на рентгенограмме признаками
- аномалии строения турецкого седла
- срастанием нескольких шейных позвонков
- уменьшение общего числа шейных позвонков
051. Аномалией Арнольда-Киари называется патология, при которой имеется
- сращение шейных позвонков
- сращение 1-го шейного позвонка с затылочной костью
+ смещение вниз миндаликов мозжечка
- расщепление дужки 2-го шейного позвонка
- расщепление дужки 1-го шейного позвонка
052. Наиболее информативными методами исследования при врожденной аномалии мозга Денди-Уолкера являются
+ КТ, МРТ головного мозга
- рентгенография кранио-вертебрального перехода
053. Клиническая картина врожденной юношеской торсионной дистонии (форма Сегавы) отличается наличием
- синдрома сенситивной атаксии
- синдром нарушения чувствительности
054. Клиническая картина ювенильной формы и формы Вестфаля при хорее Гентингтона, кроме хореического гиперкинеза, включает
- ничего из вышеперечисленного
055. При лечении типичной формы хореи Гентингтона обычно применяют
056. Мозжечковую диссинергию Ханта от миоклонус-эпилепии Унферрихта-Лундборга отличает
- наличие мозжечковых синдромов
+ отсутствие экстрапирамидных симптомов
- отсутствие пирамидных симптомов
- наличие нарушений поверхностной чувствительности
- отсутствие нарушений глубокой чувствительности
057. Клиническая картина миоклонус-эпилепсии Унферрихта-Лундборга, кроме характерных миоклоний и судорожных приступов, включает
+ экстрапирамидную ригидность и снижение интеллекта
058. Миоклониеские гиперкинезы при миоклонус-эпилепсии Унферрихта-Лундборга усиливаются
- при эмоциональном стрессе
- при внезапных сенсорных раздражениях
- при закрывании глаз
- ничего из вышеперечисленного
059. При болезни Фридрейха имеет место
+ рецессивный тип наследования
- доминантный тип наследования
- сцепленный с полом (через Х-хромосому)
- сцепленный с полом (через Y-хромосому)
060. Среди спиноцеребеллярных атаксий болезнь Фридрейха отличается наличием
- поражением мышцы сердца
- ничего из вышеперечисленного
061. Мозжечковая атаксия Пьера-Мари отличается от атаксии Фридрейха
- наличием пирамидных патологических симптомов
- наличием глазодвигательных нарушений
- высокими сухожильными и периостальными рефлексами
- ничем из вышеперечисленного
062. Для семейной спастической параплегии (болезни Штрюмпеля) характерно преобладающее поражение следующих спинальных анатомических структур
- клеток передних рогов
- задних канатиков спинного мозга
063. Характерной чертой нижнего парапареза при болезни Штрюмпеля является
- преобладание слабости над спастичностью
+ преобладание спастичности над слабостью
- преобладание мозжечковых симптомов над пирамидными
- сочетание пирамидных симптомов с фибрилляцией мышц
064. Тип наследственности при спастической семейной параплегии (болезни Штрюмпеля) характеризуется как
- рецессивный, сцепленный с полом (через Х-хромосому)
065. Нейрофибромы при болезни Реклингаузена могут локализоваться
- по ходу периферических нервов
- в спинномозговом канале по ходу корешков
- интракраниально по ходу черепных нервов
+ на любом из указанных участков
066. Тип наследования нейрофиброматоза (болезни Реклингаузен- характеризуется как
- рецессивный, сцепленный с полом (через Х-хромосому)
- ничего из перечисленного не верно
067. Для подтверждения интракраниального поражения при энцефалотригеминальном ангиоматозе достаточно произвести
+ рентгенокраниографию, КТ, МРТ головы
068. Тип наследования атаксии-телеангиэктазии (синдром Луи-Бар) характеризуется как
- рецессивный, сцепленный с полом (через Х-хромосому)
9) НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ НЕРВНОЙ СИСТЕМЫ
001.Основными задачами медицинской генетики является изучение
а)законов наследственности и изменчивости человеческого организма
б)популяционной статистики наследственных заболеваний
в)молекулярных и биохимических аспектов наследственности
г)изменения наследственности под воздействием факторов окружающей среды
002.Доминантный признак по закону Менделя проявляется при скрещивании во втором поколении с частотой
003.Доминантный ген - это ген, действие которого
а)выявляется в гетерозиготном состоянии
б)выявляется в гомозиготном состоянии
в)выявляется в гетеро- и гомозиготном состоянии
г)неверно все из перечисленного
004.Генотип организма представляет собой систему взаимодействия генов, при которой наследственные признаки определяются путем участия
а)одного гена в определении одного признака
б)одного гена в определении многих признаков
в)многих генов в определении одного признака
д)верно все перечисленное
а)здорового носителя мутантного гена
б)больного носителя мутантного гена
в)здорового родителя больного с признаками наследственного заболевания
г)ребенка, больного наследственным заболеванием
а)здорового родителя больного наследственным заболеванием
б)ребенка больного наследственным заболеванием
в)родного брата или сестру (но не близнецов) больного наследственным заболеванием
007.Фенотип - это совокупность признаков и свойств организма, проявление которых обусловлено
а)действием доминантного гена
б)действием рецессивного гена
в)взаимодействием генотипа с факторами среды
008.Кариотип - это совокупность особенностей хромосомного набора (комплекса) клетки, определяющаяся
а)числом половых хромосом
009.Аутосомно-доминантный тип наследования отличается
а)преимущественным поражением лиц мужского пола
б)преобладанием в поколении больных членов семьи
в)проявлением патологического наследуемого признака во всех поколениях без пропуска
010.Аутосомно-рецессивный тип наследования отличается тем, что
а)соотношение здоровых и больных членов семьи равно 1:1
б)заболевание не связано с кровным родством
в)родители первого выявленного больного клинически здоровы
011.Рецессивный тип наследования, связанный с Х-хромосомой (сцепленный с полом), отличается тем, что
а)соотношение больных мужчин в каждом поколении равно 2:1
б)заболевают только мужчины
в)заболевают только женщины
г)признаки болезни обязательно находят у матери пробанда
д)неверно все перечисленное
012.Причиной хромосомных заболеваний могут быть
а)изменения числа хромосом
б)изменения размера хромосом
в)нарушения структуры хромосом
г)влияние факторов внешней среды
013.Фенотипическими признаками хромосомных болезней являются
а)нарушения психического развития
б)нарушения физического развития
в)множественные пороки развития
014.Индуцированный мутагенез вызывают следующие факторы
д)все перечисленные факторы
015.В основу классификации наследственных болезней, учитывающей их генетическую природу, положены особенности
в)количественных изменений хромосом
016.Основным биохимическим признаком фенилкетонурии является повышение содержания
017.Для клинических проявлений гликогеновой миопатии (болезнь Мак-Ардля) является характерным наличие
а)болезненных пароксизмов в мышцах
б)патологической мышечной утомляемости
в)псевдогипертрофии мышц голеней
018.При поздней форме амавротической идиопатии Куфса у взрослых наблюдают
019.Нарушение движений при ювенильной форме амавротической идиотии Баттена - Шпильмейра - Фогта обусловлено поражением
д)всего перечисленного, кроме в)
е)всего перечисленного, кроме г)
020.Поражение нервной системы при лейкодистрофии происходит в результате
а)избыточного накопления липидов в нервных клетках
б)утраты липидов нервными клетками
в)распада липидов миелина и накопления продуктов распада в центральной нервной системе
021.Для порфирии является характерным наличие
в)порфобилиногена в моче
022.Прогрессирующие мышечные дистрофии обусловлены поражением
а)цереброспинальных пирамидных путей
б)мотонейронов передних рогов спинного мозга
в)периферического двигательного нейрона
е)ничего из перечисленного
023.Спинальная амиотрофия Верднига - Гоффмана наследуется
а)по аутосомно-доминантному типу
б)по аутосомно-рецессивному типу
в)по рецессивному типу, связанному с полом (Х-хромосома)
г)по доминантному типу, связанному с полом
024.Изменение контура ног по типу "опрокинутой бутылки" обусловлено изменением массы мышц
а)при амиотрофии Шарко - Мари - Тута
б)при гипертрофической невропатии Дежерина - Сотта
в)при мышечной дистрофии Эрба
г)при мышечной дистрофии Беккера - Киннера
д)при амиотрофии Кугельберга - Веландера
025.Амиотрофия Шарко - Мари - Тута обусловлена первичным поражением
а)передних рогов спинного мозга
б)периферических двигательных нервов
в)мышц дистальных отделов конечностей
026.Тип наследования при амиотрофии Шарко - Мари - Тута характеризуется как
в)сцепленный с полом (через Х-хромосому)
д)ничего из перечисленного
027.Прогрессирующая мышечная дистрофия формы Ландузи - Дежерина наследуется
а)по аутосомно-доминантному типу
б)по аутосомно-рецессивному типу
в)по рецессивному типу, сцепленному с полом (через Х-хромосому)
г)по всему перечисленному
028.Псевдогипертрофии наблюдают при следующих формах прогрессирующей мышечной дистрофии
б)тип Беккера - Киннера
в)тип Ландузи - Дежерина
029.Тип наследования при миопатии Томсена характеризуется как
в)сцепленный с полом (через Х-хромосому)
д)ничего из перечисленного
030.При атрофической миотонии преобладает слабость мышц
б)проксимальных отделов конечностей
в)дистальных отделов конечностей
031.Тип наследования при атрофической миотонии Штейнерта - Баттена характеризуется как
в)сцепленный с полом (через Х-хромосому)
д)ничего из перечисленного
032.Тип наследования при гиперкалиемическом периодическом параличе характеризуется как
в)сцепленный с полом (через Х-хромосому)
033.Тип наследования при гипокалиемическом периодическом параличе характеризуется как
в)сцепленный с полом (через Х-хромосому)
034.Нарушения медно-белкового обмена при гепатоцеребральной дистрофии Вильсона - Коновалова обусловлены дефектом гена следующей хромосомы
035.Исследование плазмы больного гепатоцеребральной дистрофией выявляет
а)повышение уровня церулоплазмина и гиперкупремию
б)понижение уровня церулоплазмина и гиперкупремию
в)повышение уровня церулоплазмина и гипокупремию
г)понижение уровня церулоплазмина и гипокупремию
036.При дрожательной и дрожательно-ригидной форме гепатоцеребральной дистрофии Вильсона - Коновалова преобладает тремор
а)покоя в кистях
б)интенционный в руках
в)хлопающий в руках
г)статодинамический в туловище
037.Тип наследования при гепатоцеребральной дистрофии характеризуется как
в)рецессивный, сцепленный с полом (через Х-хромосому)
038.Приступ пароксизмальной миоплегии при гипокалиемической форме болезни Вестфаля - Шахновича обычно возникает
а)во время тяжелой физической нагрузки
б)сразу после тяжелой физической нагрузки
в)в состоянии полного покоя днем
г)во время ночного сна
д)во всех перечисленных состояниях
039.Приступ миоплегии при гиперкалиемической (болезнь Гармсторпа) и нормокалиемической форме (болезнь Посканцера и Керра) возникает
а)во время тяжелой физической нагрузки
б)во время отдыха после физической нагрузки
в)в состоянии покоя днем
г)во время ночного сна
040.Тип наследования при хорее Гентингтона характеризуется как
в)рецессивный, сцепленный с полом (через Х-хромосому)
041.Клиническая картина типичной хореи Гентингтона, кроме хореического гиперкинеза, включает
а)пластическую экстрапирамидную ригидность
б)симптом "зубчатого колеса"
042.Нейрохимические изменения в подкорковых ядрах при болезни Паркинсона характеризуются следующими изменениями моноаминов мозга
б)увеличением содержания ацетилхолина
в)снижением содержания норадреналина
043.Болезнь Паркинсона может проявиться следующими синдромами
044.К дофасодержащим препаратам для лечения болезни Паркинсона относятся
045.При наследственном эссенциальном дрожании тремор обычно имеет следующий характер
046.При лечении болезни Паркинсона ежедневная доза L-допа не должна превышать
047.При лечении болезни Паркинсона дофасодержащими средствами неврологические побочные симптомы проявляются
048.При лечении болезни Паркинсона холинолитиками (циклодол, норакин) побочные симптомы проявляются
б)двоением в глазах
г)сухостью во рту
049.Лечение холинолитиками болезни Паркинсона противопоказано, если у больного имеются
д)любые из перечисленных заболеваний
050.Лечение холинолитиками болезни Паркинсона противопоказано при заболевании
д)при всех перечисленных заболеваниях
051.При комбинированном лечении болезни Паркинсона витамин В6 уменьшает эффективность следующих препаратов
052.При эссенциальном наследственном дрожании препаратами выбора являются
а)a-адренергические блокаторы (пирроксан)
б)b-адренергические блокаторы (анаприлин)
в)дофасодержащие средства (L-допа, наком)
г)агонисты дофаминовых рецепторов (бромкриптин)
е)все перечисленные препараты
053.Клиническая картина врожденной юношеской торзионной дистонии (форма Сегава) отличается наличием
в)синдрома сенситивной атаксии
054.При лечении типичной формы хореи Гентингтона обычно применяют
055.Мозжечковую диссинергию Ханта от миоклонус-эпилепсии Унферрихта - Лундборга отличает
а)наличие мозжечковых симптомов
б)отсутствие пирамидных симптомов
в)отсутствие экстрапирамидных симптомов
г)отсутствие нарушений глубокой чувствительности
056.Клиническая картина миоклонус-эпилепсии Унферрихта - Лундборга, кроме характерных миоклоний и судорожных приступов, включает
057.Миоклонические гиперкинезы при миоклонус-эпилепсии Унферрихта - Лундборга усиливаются
а)при эмоциональном стрессе
б)при внезапных сенсорных раздражениях
в)при закрывании глаз
д)при всем перечисленном
058.При болезни Фридрейха имеет место
а)рецессивный тип наследования
б)доминантный тип наследования
в)сцепленный с полом (через Х-хромосому)
059.Среди спиноцеребеллярных атаксий болезнь Фридрейха отличается наличием
в)поражением мышцы сердца
г)снижением или выпадением рефлексов
060.Мозжечковая атаксия Пьера - Мари отличается от атаксии Фридрейха
а)наличием пирамидных патологических симптомов
б)наличием глазодвигательных нарушений
061.Для семейной спастической параплегии (болезни Штрюмпеля) характерно преобладающее поражение следующих спинальных анатомических структур
в)клеток передних рогов
г)задних канатиков спинного мозга
062.Характерной чертой нижнего парапареза при болезни Штрюмпеля является
а)преобладание слабости над спастичностью
б)преобладание спастичности над слабостью
в)преобладание мозжечковых симптомов над пирамидными
г)сочетание пирамидных симптомов с фибрилляцией мышц
д)сочетание пирамидных симптомов с сенситивной атаксией
063.Тип наследственности при спастической семейной параплегии (болезни Штрюмпеля) характеризуется как
в)рецессивный, сцепленный с полом (через Х-хромосому)
064.Нейрофибромы при болезни Реклингаузена могут локализоваться
а)по ходу периферических нервов
б)в спинномозговом канале по ходу корешков
в)интракраниально по ходу черепных нервов
г)на любом из указанных участков
065.Тип наследования нейрофиброматоза (болезни Реклингаузена) характеризуется как
в)рецессивный, сцепленный с полом (через Х-хромосому)
д)неверно все перечисленное
066.Интракраниальный ангиоматоз при синдроме Стерджа - Вебера поражает
г)одинаково часто все перечисленные структуры
067.Для подтверждения интракраниального поражения при энцефалотригеминальном ангиоматозе достаточно произвести
068.Тип наследования атаксии-телеангиэктазии (синдром Луи - Бар) характеризуется как
в)рецессивный, сцепленный с полом (через Х-хромосому)
069.Для болезни Дауна характерно сочетание следующих признаков
а)округлый череп, готическое небо, синдактилия, гипотония мышц
б)долихоцефалия, расщепление неба, арахнодактилия, гипертонус мышц
в)краниостенотический череп, заячья губа, наличие 6-го пальца, хореоатетоз
г)наблюдается сочетание любых перечисленных признаков
070.Тип наследования при синдроме Шегрена - Ларссена характеризуется как
в)рецессивный, сцепленный с полом (через Х-хромосому)
071.Достаточными клиническими признаками в диагностике сирингомиелии являются
а)сегментарные диссоциированные нарушения чувствительности
б)наличие дизрафических черт строения опорно-двигательного аппарата
в)прогрессирующая атрофия мышц в участках, соответствующих сегментарным нарушениям чувствительности
г)нижний спастический парапарез
072.Для переднероговой формы сирингомиелии характерны
а)нарушения проприоцептивной чувствительности
в)диссоциированный тип нарушений чувствительности
е)ничего из перечисленного
073.Синдром Клиппеля - Фейля характеризуется на рентгенограмме признаками
в)остеопороза турецкого седла
г)выступанием зуба второго шейного позвонка в область проекции задней черепной ямки
д)срастанием нескольких шейных позвонков
074.Аномалией Арнольда - Киари называется патология, при которой имеется
а)сращение шейных позвонков
б)сращение 1-го шейного позвонка с затылочной костью
в)смещение вниз миндаликов мозжечка
г)расщепление дужки 1-го шейного позвонка
075.Наиболее информативными методами исследования при врожденной аномалии мозга Денди - Уолкера являются
б)компьютерная томография мозга
г)рентгенография кранио-вертебрального перехода
Читайте также:
