
Атаксия при эпилепсии у детей
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ
В 1968 г. May и White сообщили о семье, в которой у 6 больных в четырех поколениях был обнаружен синдром, включающий миоклонус, мозжечковую атаксию и пейросепсорную глухоту.
Клинические данные. Нервная система. У 4 из 6 больных в анамнезе имелись указания на большие судорожные припадки и у 2 из этих 4 наблюдался миоклонус. Миоклонические эпизоды появлялись в возрасте между 12 и 20 годами и заключались в сокращениях мускулатуры головы, туловища и конечностей. Некоторое время спустя, обычно в третьем десятилетии жизни, начинались генерализованные припадки. У 2 из 6 больных больших судорожных припадков не было. В юности появлялись нарушения координации в руках и ногах.
Они наблюдались у 3 из 6 больных. Атаксия, когда она имелась, прогрессировала медленно. Неврологическое исследование 3 больных выявило неустойчивую походку на широко расставленных ногах, легкий или умеренный интенционный тремор, тремор при пальценосовой или пяточно-коленной пробах, атаксию туловища. Нистагма, патологии черепно-мозговых нервов, потери чувствительности и мышечных атрофии не выявлено, хотя речь у больных с атаксией была дизартричной. Психических расстройств не отмечалось.
Орган слуха. Нарушения слуха впервые были замечены в детстве или в молодости. Они медленно прогрессировали, приводя в результате к резко выраженной глухоте в поздние годы жизни. На аудиограмме одного больного 32 лет была обнаружена двусторонняя нейросенсорная глухота (около 40 дБ). У другого больного в 50-летнем возрасте отмечалась глубокая потеря звуковоеприятия. Он слышал только громкие звуки, направленные в ухо.
Вестибулярная система. Результаты вестибулярных проб не описаны.
Лабораторные данные. У 3 обследованных больных на электроэнцефалограмме обнаружена нормальная активность в покое. У 2 больных с эпилептическими припадками в анамнезе световая стимуляция индуцировала продолжительные генерализованные спайки. В одном случае наблюдалось сочетание мноклонических сокращений в двуглавой мышце с корковыми спайками.
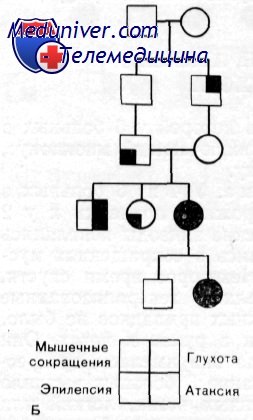
Наследственность. Проявления синдрома у 6 больных умеренно варьировали. У пробанда и у его матери был полный синдром, включающий глухоту, миоклонус, эпилепсию и атаксию. У дяди по линии матери отмечались глухота и атаксия без эпилепсии. У тетки и у деда по матери была только эпилепсия, а у прадеда по матери — только глухота. Никто из этих трех последних лиц обследован не был. Создается впечатление, что синдром наследуется по аутосомно-доминантному типу с варьирующей экспрессивностью.
Диагноз. Миоклонус характерен для нескольких разных заболеваний. Доброкачественный эссенциальный миоклонус наследуется по аутосомно-доминантному типу, но он не сочетается с атаксией или глухотой. Миоклоническая эпилепсия Лафора наследуется также по аутосомно-доминантному типу, но она характеризуется быстро нарастающим распадом психики, развивающимся в середине подросткового возраста после выявления эпилепсии.
При аутосомно-рецессивно наследующейся миоклонической эпилепсии Унверрихта, после выявления симптомов болезни, в юности развивается только легкое слабоумие. Миоклоническая эпилепсия наблюдается при гаргоилизме, который наследуется по аутосомно-рецессивному типу, выявляется в первые несколько лет жизни и приводит к смерти в 10-летнем возрасте. Миоклоническая эпилепсия Рамсея Ханта (Ramsey Hunt) наследуется также по аутосомно-рецсссивпому типу и включает миоклонус с атаксией, но без глухоты.
Лечение. При мноклопических сокращениях наиболее эффективен диазепам (диазепам, валиум). Большие судорожные припадки эффективно лечатся фенитоином (фенитоин, дифенилгидантоин, дилантин) и фенобарбиталом. При дефектах слуха можно использовать слуховые аппараты.
Прогноз. Заболевание медленно прогрессирует и ведет к расстройствам походки и слуха.
Выводы. Характеристика данного синдрома включает: 1) аутосомно-доминантное наследование с варьирующей экспрессивностью; 2) миоклоническую и grand mal эпилепсию, начинающуюся между 12 и 30 годами и встречающуюся примерно в половине случаев; 3) медленно прогрессирующую мозжечковую атаксию, начинающуюся в юности; 4) медленно прогрессирующую нейросенсорную глухоту, начинающуюся в детстве.
Медицинский эксперт статьи

Прогрессирующая миоклонус-эпилепсия относится к полиэтиологическим синдромам. В настоящее время выделено около 15 нозологических форм, сочетающихся с прогрессирующей миоклонус-эпилепсией. Прогрессирующей миоклонус-эпилепсией называют сложный синдром, включающий сочетание миоклонуса, эпилепсии, когнитивных нарушений и различных других неврологических нарушений (чаще всего мозжечковой атаксии) с прогрессирующим течением.
Диагностическая триада прогрессирующей миоклонус-эпилепсии:
- Миоклонические припадки.
- Тонико-клонические судорожные припадки.
- Прогрессирующие неврологические расстройства (обычно атаксия и деменция).

[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Заболевания, при которых встречается прогрессирующая миоклонус-эпилепсия
Прогрессирующая миоклонус-эпилепсия встречается при следующих заболеваниях:
Болезни, пограничные с прогрессирующей миоклонус - эпилепсией (сочетание эпилепсии и миоклонуса):
- Сочетание первичной эпилепсии и семейных миоклоний (редко)
- Болезнь Тея-Сакса (Tay-Sachs)
- Фенилкетонурия
- Липофусциноз новорожденных (синдром Santavuori-Haltia)
- Подострый склерозирующий панэнцефалит
- Болезнь Вильсона-Коновалова
- Болезнь Крейтцфельдта-Якоба
Острые состояния, при которых возможно появление миоклонус-эпилепсии:
- Интоксикация метилбромидом, висмутом, стрихнином.
- Вирусные энцефалиты.
[7], [8], [9], [10], [11], [12], [13]
Это заболевание описано в двух подгруппах больных. Одна форма выявлена впервые в Финдляндии и была названа впоследствии балтийским миоклонусом. Другая - на юге Франции (Марсель) и называется в настоящее время средиземноморским миоклонусом.
Диагностические критерии болезни Унферрихта-Лундборга включают:
- Начало болезни в возрасте между 6 и 15 годами(в 86 % случаев - между 9 и 13 годами).
- Тонико-клонические эпилептические припадки.
- Миоклонус.
- ЭЭГ: пароксизмы спайков или комплексов полиспайк-волна с частотой 3-5 в сек.
- Прогрессирующее течение с присоединением грубой мозжечковой атаксии и деменции.
Миоклонус при болезни Унферрихта-Лундборга, как и при всех прогрессирующих миоклонус-эпилепсиях, относится к корковому миоклонусу. Он может быть как спонтанным и наблюдаться в покое, так и связанным с движениями (акционный миоклонус или миоклонус действия) и тем самым существенно затруднять повседневную активность больного. Миоклонические подёргивания провоцируются также сенсорными стимулами (стимул-сенситивный или рефлекторный миоклонус) такими как прикосновение, свет, звук и др. Миоклонус может иметь разное распределение по телу и вариирует по интенсивности даже у одного и того же больного. Обычно он асинхронный, может преобладать в одной конечности или одной половине тела, при усилении он может распространяться на другие части тела и иногда протекает в виде генерализованного миоклонического приступа без или с минимальным нарушением сознания. У большинства больных миоклонус имеет прогрессирующее течение.
У большинства пациентов развивается выраженная мозжечковая атаксия и деменция.
У больных средиземноморским миоклонусом (то, что раньше называли синдромом Рамсея Ханта) эпилептические припадки и деменция выражены весьма слабо и в отдельных случаях могут даже отсутствовать. Отвественный ген при болезни Унферрихта-Лундберга расположен на 21 хромосоме, что было подтверждено у больных со средиземноморским вариантом болезни.
Заболевание наследуется по аутосомно-рецессивному типу и начинается в возрасте 6-19 лет. Манифестным проявлением являются генерализованные тонико-клонические эпилептические припадки. Последние часто сочетаются с парциальными затылочными пароксизмами в виде простых галлюцинаций, скотом или более сложных зрительных расстройств. Зрительные пароксизмы - характерный признак болезни Лафора, наблюдаемый у 50 % больных уже на ранних стадиях заболевания. Вслед за эпилептическими приступами обычно развивается тяжёлый миоклонус покоя и действия. Атаксия нередко замаскирована тяжёлым миоклонусом. Нарушения когнитивных функций могут проявляться уже в дебюте болезни. Более грубые психические нарушения характерны для развёрнутой стадии заболевания. Возможно преходящая корковая слепота. В терминальной стадии больные прикованы к постели, у них отмечается деменция. Летальный исход наступает через 2-10 лет от начала заболевания.
Диагноз. При световой микроскопии обнаруживаются тельца Лафора в коре мозга, ткани печени и скелетных мышцах. Наиболее информативным и доступным методом является исследование биоптатов кожи, особенно в области предплечья.
[14]
Цероидный липофусциноз (церебро-ретинальные дегенерации) относится к липидозам и характеризуется отложением аутофлюоресцентных липопигментов в центральной нервной системе, гепатоцитах, сердечной мышце, сетчатке. Первичный биохимический дефект, лежащий в основе заболевания, неизвестен. Цероидный липофусциноз является одной из причин прогрессирующей миоклонус-эпилепсии. Выделяют несколько типов цероидного липофусциноза: инфантильный, поздний инфантильный, ранний ювенильный или промежуточный, ювенильный, форма взрослых.
Инфантильный тип Сантавуори-Халтиа манифестирует после 6-8 мес. и в строгом смысле не относится к прогрессирующим миоклонус-эпилепсиям.
[15], [16], [17], [18], [19], [20], [21]
[22], [C. - PubMed - NCBI" target="_blank" rel="noopener noreferrer">23]
Болезнь Гоше (Gaucher) известна в трёх формах: инфантильной (тип I), ювенильной (тип II) и хронической (тип III). Последний тип болезни Гоше может проявляться прогрессирующей миоклонус-эпилепсией. Заболевание обусловлено недостаточностью бета-глюкоцереброзидазы и характеризуется накоплением глюкоцереброзида в различных тканях организма.
[24], [25]
[26], [27], [28], [29]
Симптоматическая эпилепсия - заболевание нервной системы органического генеза, возникающее преимущественно в детском возрасте. Эта болезнь требует постоянного наблюдения, ухода за пациентом и длительной терапии для улучшения состояния человека. Диагностика заболевания осуществляется при помощи эпилептолога и невролога на основании жалоб и результатов проведенного обследования. Лечение проводится при помощи средств, позволяющих снять выраженность симптомов.
- 1. Симптоматическая эпилепсия: описание заболевания
- 2. Основные клинические проявления
- 2.1. Формы
- 3. Диагностика
- 4. Лечение
Симптоматическая (вторичная) эпилепсия - одна из форм заболеваний нервной системы, которая характеризуется появлением эпилептических полиморфных приступов. Эта патология преимущественно возникает у детей, однако бывают случаи ее развития у взрослых людей. Причиной этого недуга являются черепно-мозговые травмы, доброкачественные и злокачественные опухоли, инсульты и врожденные аномалии функционирования головного мозга.
Основной фактор развития этого заболевания - нарушение обменных процессов в нервных клетках. Эта патология возникает по причине преобладания возбуждения над торможением в головном мозге. Инфекции плода и родовые травмы влияют на развитие этой патологии. К причинам возникновения симптоматической эпилепсии также можно отнести:
- сахарный диабет;
- заболевания печени и почек;
- рассеянный склероз;
- нарушения мозгового кровообращения;
- инсульт;
- ревматизм;
- инфекционные заболевания головного мозга (абсцесс, энцефалит, менингит).

Основным признаком вторичного генерализованного приступа являются судорожные припадки. Отмечаются двигательные нарушения: атаксия (сбой согласованности различных мышц при отсутствии мышечной слабости у пациента), парезы или параличи. Наблюдаются задержка психомоторного развития и возникновение умственной отсталости (олигофрении).
У таких пациентов отмечаются вегетативные нарушения. Они проявляются повышенным слюно- и потоотделением. Отмечаются жалобы на учащенное сердцебиение (тахикардию) и затрудненность дыхания.

Существует несколько форм фокальной эпилепсии, каждая из которых отличается симптомами и их выраженностью:
| Вид | Характеристика |
| Височная | Отмечаются нарушения памяти, зрительные и слуховые галлюцинации. Перед началом приступа возникает выключение сознания. При обследовании с помощью ЭЭГ (электроэнцефалографии) нарушений не выявляется |
| Теменная | Нарушения сексуального поведения. Отмечаются патологии чувствительности (болевые и температурные) |
| Затылочная | Визуальные галлюцинации, патологии зрительной системы. Наблюдаются болевые ощущения и дискомфорт в области глазных яблок |
| Лобная | Возникает сомнамбулизм (лунатизм) и сноговорение. Наблюдаются нарушения поведения и двигательной активности. Отмечается быстрый переход припадков в сложную форму |
У некоторых детей развивается особая разновидность симптоматической эпилепсии, называемая синдромом Веста. Этой патологией страдают преимущественно мальчики в возрасте от 3 до 8 месяцев. У таких пациентов наблюдаются частые судорожные приступы, которые можно увидеть на ЭЭГ, и нарушения психомоторного развития.
Это заболевание почти не поддается лечению. Наблюдаются наклоны тела вперед. Отмечаются судороги верхних и нижних конечностей. Приступы длятся несколько секунд, а затем наступает небольшая пауза. При припадке у пациентов закатываются глаза. Тело выгибается.

Наблюдается снижение интеллекта. Анализ МРТ позволяет разграничить симптоматическую эпилепсию от опухолей головного мозга, заболеваний сосудов и рассеянного склероза. ЭЭГ дает возможность выявить электрическую активность мозга, которая характерна для эпилептических приступов. Благодаря этому методу удается определить локализацию и форму заболевания в зависимости от того, в какой области мозга зафиксирован очаг активности.
Осмотр невролога начинается со сбора анамнестических сведений, который включает жалобы и заболевания. Необходимо установить возраст начала проявлений и возможные причины возникновения этой патологии. Для исключения других соматических заболеваний нужно пройти осмотр офтальмолога, эндокринолога и врача-генетика.
Самыми важными показателями эффективности лечения являются снижение количества приступов и улучшения на ЭЭГ. Курс терапии у детей и взрослых составляет длительное время, а иногда осуществляется пожизненно. Это необходимо, чтобы сгладить проявления симптомов и уменьшить частоту возникновения.
Для лечения такой эпилепсии применяют противосудорожные препараты. Дозировка лекарств зависит от вида заболевания, выраженности проявлений и индивидуальных особенностей пациента. Лечить эту патологию следует не менее 5 лет.
При ранней отмене терапии можно спровоцировать рецидив заболевания. Если у пациента возникают какие-либо сопутствующие болезни, важно осуществить и их лечение. В противном случае это может усугубить развитие основного заболевания.
Эпизодические атаксии, то есть временно возникающая шаткость при ходьбе и хаотичные размашистые движения, относят к генетическим неврологическим заболеваниям. Пациент шатается при ходьбе, может упасть и не встать до окончания приступа. Он не способен взять предмет, застегнуть пуговицу, самостоятельно выпить воды.
Периоды относительного благополучия сменяются приступами раскоординации движений, ребенок не может некоторое время управлять своими движениями. Внешне эпизодические атаксии напоминают проявления эпилептического синдрома, но по сути имеется ряд существенных отличий:
- Во-первых, во время эпизодической атаксии пациент не теряет сознание, а ЭЭГ не регистрирует эпилептической активности.
Клинических вариантов эпилептических пароксизмов крайне много. Только мониторинг ЭЭГ позволит отдифференцировать эпилепсию от другого заболевания и вовремя выставить правильный диагноз, подобрать лечение.
- Во-вторых, атака провоцируется физиологическим или психологическим стрессом: жара или холод, усталость, перенапряжение сил, эмоционально значимые события.
Атаксия способна коснуться и речевого аппарата (отмечаются нарушения речи), тогда мы слышим монотонную, с придыханием, неожиданными паузами и эффектом стаккато, скандированную речь.
Такая разбалансированность произвольных двигательных процессов длится от нескольких минут до нескольких часов, а затем исчезает, в ряде случаев – бесследно до нового пароксизма (приступа). Иногда и между приступами сохраняется небольшая атаксия, потому что ткани мозжечка, центра координации движений, постепенно атрофируются.

Проявляется болезнь в детском или подростковом возрасте, эпизоды атаксии случаются с разной частотой: несколько раз за день, ежедневно, пару раз в неделю, ежемесячно. С возрастом этот показатель урежается.
Эпизодическая атаксия первого типа. Приступ выражен миокимией в сочетании с атаксией. Миокимия или мышечная волна представляет собой периоды сокращений мышцы или ее части, видимыми подергиваниями без совершения движения. Такая атака длится пару минут, впервые проявляясь в раннем детстве. Причиной заболевания является мутация генов, обеспечивающих калий-натриевую передачу:калиевых каналов становится меньше, форма их меняется, клеточные мембраны ощущают дефицит калия, в результате клетки медленно расслабляются после напряжения. Значительно страдает баланс возбуждения и торможения мозжечка, клеток двигательных нейронов спинного мозга и периферических нервов.
Собственно, заболевание развивается у детей среднего возраста, примерно от 7 до 12 лет. Испуг, физические нагрузки, перенесенные соматические заболевания, яркие эмоции, резкие движения – все это способно спровоцировать приступ.
Ребенок испытывает стресс, реагируя на него мышечным напряжением и попыткой действовать быстро. Однако избыточное возбуждение приводит к тому, что клетки часто напрягаются и не успевают расслабиться. В результате развиваются локальные мышечные подергивания (миокимии) и теряется контроль над движениями (атаксия). Вероятность развития эпилепсии у таких детей в десять раз выше, чем обычно.
Клиническая картина
Атаксия сопровождается мозжечковыми нарушениями:
- кружится голова (головокружение);
- двоится в глазах;
- миокимии – изолированные мышечные подергивания;
- тошнит, встречается рвота;
- нарушаются речь и зрение.
Такой приступ длится от нескольких минут до нескольких часов, повторяясь до 10-15 раз за сутки. Помимо атаксических атак в межприступный период отмечаются миокимии – подергивания мышц лица, стоп, голеней, кистей, предплечий.
Эпизоды могут возникать спонтанно и после движения, испуга, на фоне заболевания, сильных положительных и отрицательных эмоций.
Для диагностики необходимо провести генетический анализ, электромиографию, которая обнаружит постоянное напряжение отдельных мышечных волокон. На ЭЭГ при эпизодической атаксии эпи-активности как правило нет, однако во многих случаях заболевание протекает в сочетании с эпилепсией и требует проведения длительной электроэнцефалограммы бодрствования со сном.

Лечение направлено на купирование и предупреждение развития приступов. Оно включает в себя противоэпилептическую и седативную терапию (ацетазоламид, карбамазепин, фенотонин, вальпроат, фенобарбитал). Заболевание неизлечимо, но с возрастом атаки легче и реже.
Мутация гена приводит к изменению белка кальциевого канала (альфа – 1А-субъединица потенциал зависимого кальциевого канала). Этот белок входит в состав клеток мозжечка, формирует просвет каналов, находящихся в этом отделе головного мозга мозжечке, именно они участвуют и в образовании синоптической передачи импульса к нервам и мышцам. Изменения этого же гена встречаются при семейных мигренях, когда к приступам головной боли добавляется небольшая атаксия, эпизодической гемиплегии, спиноцеребеллярной атаксии. В последнем случае атаксия носит постоянный характер, постепенно увеличивая свои проявления. Встречается мутированная часть генов с 80% вероятностью среди членов одной семьи, хотя в целом эпизодическая атаксия – очень редкоезаболевание.
Волнение, физические и психологические стрессы, кофе, алкоголь или психостимулирующие средства, беременность, высокая температура тела или жара провоцируют приступы атаксии, так как на их развитие влияет гормональный фон пациента.
У мальчиков и взрослых мужчин заболевание протекает тяжелее: приступы чаще, нарушения движений, напоминающие судороги, более выражены. Развивается эпизодическая атаксия обычно в детстве, но может впервые появиться и у взрослого человека (дебют болезни от двух до тридцати с небольшим лет).
Клинические проявления:
- разлитая слабость;
- размашистые неконтролируемые движения, подобные судорожному приступу;
- иногда изменение темпа речи;
- головокружение;
- мигрень во время приступа встречается у 50% больных(сильно болит голова);
- тошнота, рвота.
Могут присоединиться следующие симптомы:
Приступ длится дольше, чем при эпизодической атаксии первого типа, от нескольких часов до 1-2 суток. Повторяются пароксизмы с различной частотой: у одних такое состояние развивается несколько раз в неделю, у других – один-два раза в год. Между атаками человек способен вести обычную жизнь, но со временем появляются проявления атаксии: пошатывание, ухудшение координации движений, нарушения баланса тела в сложных позах, а также нистагм и небольшая мышечная слабость.

Девочка пытается ответить, но во рту словно образуется каша, в глазах начинает двоиться, начинает сильно кружиться голова. Разливается невыносимая слабость, и девушка, неловко взмахнув руками, медленно падает на пол.
— Да ты пьяная! – возмущается отец, -Почему у тебя глаза бегают? Вставай немедленно!
Встать она не может – половина тело обмякает. Другой рукой она пытается опереться о ручку двери, но не может ее захватить. Голова болит и кружится, сильно тошнит.
-Что ты кричишь на ребенка? Возмущается мать. – Ты посмотри, у нее же инсульт!
Приезжает Скорая помощь, девочку несут в машину на носилках. В приемном покое голова перестает болеть, движения восстанавливаются, возвращается речь… Дается направление на консультацию к врачу-неврологу по месту жительства.
В диагностический комплекс исследований в обязательном порядке входят: мониторинг ЭЭГ (где фиксируется отсутствие эпи-активности), электромиография (где не отмечается миокимий), генетическое молекулярное исследование, нейрорадиологическое исследование головного мозга или МРТ, свидетельствующие о повреждении мозжечка.
В лечении с успехом применяют ацетазоламид, более известный как диакарб. Он хорошо снимает приступы атаксии, а на фоне постоянного приема их практически не происходит. В то же время диакарб назначается под контролем анализов мочи и крови, не применяется при беременности. К сожалению, этот препарат не влияет на межприступные проявления болезни. Можно добиться длительной ремиссии, но нистагм и шаткость могутоставаться все равно, хотя пациенты мобильны, сами передвигаются и ухаживают за собой.
В таблице №1 представлена сравнительная характеристика эпизодической атаксии первого и второго типов.
Еще реже, чем вышеописанные формы заболевания, встречаются эпизодические атаксии 3 и 4 типов. Эпизодическая атаксия третьего типа начинается в позднем подростковом периоде. Приступ сопровождается головокружением, атаксией и двоением в глазах. Со временем выраженность двигательных нарушений может увеличиваться.
Эпизодическая атаксия четвертого типа объединяет некоторые черты 1 и 2 типа болезнис одной стороны, чувствительна к диакарбу, с другой – сопровождается миокиниями. Начало болезни возможно в любом возрасте в пределах 35 лет жизни, а приступ характеризуется головокружением и непроизвольными движениями. Между атаками встречаются непроизвольные подергивания отдельных мышц, но нистагма и шаткости походки не наблюдается.
Атаксические движения не очень похожи на классический судорожный синдром, но миокимии при эпизодической атаксии первого типа напоминают эпилептический миоклонус. Поэтому для диагностики необходимы периодические ЭЭГ-исследования, тем более что для заболевания первого типа риск присоединения эпилепсии более чем высок.
Источник: глава 15, Эпизодические атаксии. Е.Д.Белоусова, А.Ю.Ермаков. М.Ю.Дорофеева и др.
Читайте также:
