
Доброкачественная инфантильная миоклоническая эпилепсия
Charlotte Dravet, Michelle Bureau
История и терминология
По нашим сведениям, на данный момент имеется 67 опубликованных в литературе случаев, из которых 10 описаны как рефлекторные (Dravet and Bureau 2002). Необходимо упомянуть о том, что в первом описании синдрома возраст дебюта приходился на период до 3 лет, в то время как последующие авторы описывали возможность и более позднего дебюта – вплоть до 4 лет 8 месяцев (Giovanardi Rossi et al 1997). Это означает, что один и тот же тип эпилепсии может дебютировать в разное время, но чаще в определенные периоды (Guerrini et al 1994).
Доброкачественная миоклоническая эпилепсия характеризуется появлением коротких миоклонических атак у здоровых младенцев в возрасте от ½ года до 3 лет. Более ранний дебют не типичен. Семейный анамнез по эпилепсии и фебрильным судорогам отягощен у 30% детей. Как правило, до появления миоклоний развитие ребенка протекает нормально, без признаков патологии. Однако имеются сообщения о наличии в анамнезе фебрильных судорог примерно у 20% случаев. Они обычно редкие, простые, как правило, предшествуют появлению миоклоний. У двоих пациентов были сопутствующие заболевания, у одного - синдром Дауна (Dravet and Bureau 2002), а у другого – диабет (Colamaria et al 1987).
В начале заболевания развитие остается нормальным, и эти движения не расцениваются родителями и педиатрами как патологические.
Интериктальная ЭЭГ может оставаться совершенно нормальной. Но миоклонии всегда сопровождаются на ЭЭГ быстрой генерализованной спайк-волновой или полиспайк-волновой активностью, частотой более 3 Гц, более или менее регулярной, длящейся от 1 до 3 секунд. Во время засыпания обычно наблюдается усиление миоклоний, которые обычно (но не всегда) прекращаются во сне. Ритмическая фотостимуляция также может провоцировать миоклонии.
Полиграфические записи демонстрируют связь между возникновением миоклоний и появлением на ЭЭГ разрядов спайк-волн или полиспайк-волн. Миоклонии короткие – от 1 до 3 секунд ) и обычно изолированные. За миоклониями может следовать короткий период атонии. Иногда после атаки может следовать произвольное движение, которое выглядит нормальным мышечным сокращением.
Интериктальная ЭЭГ детей соответствует возрасту. Спонтанные разряды спайк-волн регистрируются редко, может отмечаться медленноволновая активность над центральными областями. Если ритмическая фотостимуляция вызывает спайк-волны, последние всегда сопровождаются миоклониями. При записи сна регистрируется его нормальное разделение по фазам, во время REM-сна могут отмечаться генерализованные разряды спайк-волн.
У детей, страдающих доброкачественной миоклонической эпилепсией, другие типы приступов, в частности, абсансы или тонические приступы, не развиваются, даже если дети не получают лечение. Неврологический статус – без изменений. Интериктальный миоклонус описан только у 6 пациентов (Giovanardi Rossi et al 1997). При исследовании КТ или МРТ патологических изменений не было выявлено.
Прогноз зависит от своевременности диагностики и назначения лечения. Если не лечить, у пациента продолжаются миоклонические приступы, что может сказаться на психомоторном развитии и привести к появлению расстройств поведения. Миоклонии легко контролируются монотерапией вальпроатами, и ребенок может дальше развиваться в соответствии с возрастными нормами.
Ясного представления о механизмах развития синдрома на сегодняшний день нет. Генетика – не известна. Случаи редки сами по себе, семейных случаев не описано. Генетические связи с другими формами идиопатических генерализованных эпилепсий не установлены. Delgado-Escueta в своем исследовании не нашел ни одного случая юношеской миоклонической эпилепсии среди членов семей 24 пациентов, страдающих доброкачественной миоклонической эпилепсией младенчества (Delgado-Escueta et al 1990). Arzimanoglou описал случай заболевания второго мальчика в семье, старший брат которого страдал типичной формой миоклонических-астатических приступов, – синдромом Дузе. Этот случай поднимает вопрос о взаимосвязи двух синдромов в рамках одной большой группы идиопатических генерализованных эпилепсий детского возраста. Biondi описал случай с пациентом, у других членов семьи которого было подозрение на эпилепсию (Biondi et al 1991). У его отца и 2 сестер в ЭЭГ сна регистрировались короткие вспышки генерализованных спайк-волн.
Согласно данным немногочисленных эпидемиологических исследований, доброкачественная миоклоническая эпилепсия младенчества составляет меньше 1% всех эпилепсий (Loiseau et al 1991; Centre Saint-Paul 1997, неопубликованные данные), но около 2 % эпилепсий, начинающихся в первые 3 года жизни (Dalla Bernardina et al 1983), и 2% от всех идиопатических генерализованных эпилепсий (Centre Saint-Paul 1997, неопубликованные данные).
Нет данных о возможности профилактики синдрома.
Когда на первом году жизни у ребенка начинаются миоклонии, диагноз, который приходит на ум, это криптогенные инфантильные спазмы. Клинически спазмы отличаются от миоклоний. Они более интенсивные и вызывают отчетливое сгибание всего тела, что никогда не наблюдается при доброкачественной миоклонической эпилепсии. Помимо единичных, изолированных спазмов, у ребенка всегда наблюдаются также серии спазмов. На полиграфических записях инфантильных спазмов хорошо виден типичный паттерн короткого тонического сокращения, хорошо описанный Fusco и Vigevano, миоклонии редко носят продолжительный характер (Fusco and Vigevano 1993). Отличается и иктальная ЭЭГ – нет быстрой генерализованной спайк-волновой активности. Для нее характерны: внезапное уплощение ритма после гипсаритмии с наложением (или без) быстро-волновой активности, высокоамплитудные медленные волны, сменяемые уплощением, или даже отсутствие видимых изменений. Инфантильные спазмы всегда сопровождается изменением поведенческой активности, снижением контакта, нарушением психомоторного развития, вплоть остановки и даже регрессии. На интериктальных ЭЭГ всегда регистрируются патологические изменения – как истинная гипсаритмия, так и ее модифицированная форма, или фокальные нарушения; при этом никогда не регистрируются изолированные или короткие вспышки билатерально-синхронных спайк-волн, как при доброкачественной миоклонической эпилепсии.
В случаях, когда после нескольких обследований и психомоторное развитие, и ЭЭГ (как во время сна, так и бодрствования) остаются нормальными, даже если приступы напоминают инфантильные спазмы, то можно предположить у ребенка доброкачественный неэпилептический миоклонус (Lombroso and Fejerman 1977). У этих пациентов и в иктальной ЭЭГ нет изменений (Dravet et al 1986; Pachatz et al 1999).
На первом году жизни может дебютировать также тяжелая миоклоническая эпилепсия младенцев, но она всегда начинается с продолжительных, повторяющихся фебрильных судорог (но не с изолированных миоклоний), при этой форме всегда страдает психомоторное развитие (Dravet and Bureau 2002).
Наконец, необходимо исключать и другие эпилепсии, которые могут дебютировать в первые 3 года жизни и проявляющиеся главным образом миоклониями, имеют при этом различный прогноз. Они представляют собой различные сочетания других типов приступов, с фокальными измененими на ЭЭГ, задержкой психомоторного развития, плохим ответом на терапию АЭП и неоднозначным прогнозом (Dravet 1990).
Диагностический алгоритм довольно простой. Необходим качественный сбор анамнеза и повторные записи полиграфической видео-ЭЭГ для того, чтобы доказать наличие миоклонических атак с генерализованными разрядами спайк-волн. Миоклонии либо спонтанные, либо возникают в ответ на звук, прикосновение или ритмическую фотостимуляцию, а также при засыпании. Во время ЭЭГ сна можно увидеть некоторую активацию разрядов без изменения их морфологии, появление быстрых ритмов и фокальных нарушений. Нейровизуализация полезна (но необязательна), чтобы подтвердить отсутствие структурных нарушений головного мозга. Нейропсихологическое тестирование необходимо для проверки отсутствия нарушений психомоторного развития.
По определению прогноз благоприятный, миоклонические приступы прекращаются при условии назначения соответствующего лечения – монотерапии вальпроатами. В одном из исследований сообщалось, что только 5 пациентам было необходимо добавление второго препарата для достижения контроля над приступами (Giovanardi Rossi et al 1997). Имеющиеся данные по длительности наблюдения различны – от 9 месяцев до 27 лет. У 10 пациентов появились редкие генерализованные тонико-клонические судороги, не сочетаясь с миоклониями, из них у 3 они возникли после отмены препарата, у остальных – в подростковом возрасте (Dravet and Bureau 2002). Приступы, провоцируемые звуком или прикосновением, легче поддаются контролю, чем спонтанные. И наоборот, фотосенситивность труднее контролируется, и может регистрироваться на протяжении нескольких лет после прекращения приступов.
Прогнозировать исход по психическому развитию сложнее. В большинстве случаев прогноз довольно благоприятный. Однако в рамках продолжительных исследований описаны 12 пациентов, у которых отмечались умеренная задержка психического развития, личностные расстройства или легкие нарушения поведения (Colamaria et al 1987; Todt and Muller 1992; Giovanardi Rossi et al 1997; Dravet and Bureau 2002). Ни один из этих пациентов не был госпитализирован для специализированного лечения. Благоприятный прогноз по психологическим и когнитивным функциям также зависит от своевременности диагноза, назначения адекватного лечения и помощи со стороны родственников. Но существуют и противоположные факторы, как то - проблемы в семье и неблагоприятные особенности взаимоотношения между матерью и ребенком.
В первую очередь назначается монотерапия вальпроатами, лучше в инъекциях, т.к. дети могут отказываться пить сироп. Необходимо тщательно контролировать его уровень в плазме крови, поскольку нерегулярность приема может привести к возобновлению приступов и имитировать резистентную форму. Суточная дозировка 30 мг/кг, как правило бывает достаточной, но иногда необходимо и увеличивать дозу (Lin et al 1998).. Вальпроат также эффективен против фебрильных судорог. Если миоклонии не уходят полностью на вальпроате, можно попробовать добавить бензодиазепин (клобазам или нитразепам) или этосуксимид, либо пересмотреть диагноз. Терапию, если она переносится хорошо, необходимо продолжать в течение 3-4 лет с начала приступов, более продолжительно в случаях с фотосенситивностью. Если приступы носят исключительно рефлекторный характер, можно обойтись без приема вальпроата либо прекратить терапию раньше. В случае возникновения одного генерализованного тонико-клонического приступа в подростковом возрасте, возможно, потребуется еще один, короткий курс лечения.
Список литературы
Arzimanoglou A, Prudent M, Salefranque F. Epilepsie myoclono-astatique et epilepsie myoclonique benigne du nourrisson dans une meme famille: quelques reflexions sur la classification des epilepsies. Epilepsies 1996;8:307-15.**
Beaumanoir A, Blume W. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet Ch, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey Ltd, 2002:113-35.
Biondi R, Sofia V, Tarascone M, Leocata R. Epilessia mioclonica benigna dell’infanzia: contibuto clinico. Boll Lega Ut Epil 1991;74:93-4.
Colamaria V, Andrighetto G, Pinelli L, Olivieri A, Alfieri P, Dalla Bernardina B. Iperinsulinismo, ipoglicemia ed epilessia mioclonica benigna del lattante. Boll Lega It Epi 1987;58/59:231-3.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
Dalla Bernardina B, Colamaria V, Capovilla G, Bondavalli S. Nosological classification of epilepsies in the first three years of life. In: Nistico G, Di Perri R, Meinardi H, editors. Epilepsy: an update on research and therapy. New-York: Alan Liss, 1983:165-83.
Delgado-Escueta AV, Greenberg D, Weissbecker K, et al. Gene mapping in the idiopathic generalized epilepsies: juvenile myoclonic epilepsy, childhood absence epilepsy, epilepsy with grand mal seizures, and early childhood myoclonic epilepsy. Epilepsia 1990;31(suppl 3):S19-29.
Doose H. Myoclonic astatic epilepsy of early childhood. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence, 2nd ed. London: John Libbey Eurotext Ltd., 1992:103-14.
Dravet C. Les epilepsies myocloniques benignes du nourrisson. Epilepsies 1990;2:95-101.
Dravet C, Bureau M. L’epilepsie myoclonique benigne du nourrisson. Rev Electroenceph Neurophysiol Clin 1981;11:438-44.
Dravet C, Bureau M, Giraud N, Roger J, Gobbi G, Dalla Bernardina B. Benign myoclonus of early infancy or benign non-epileptic spasms. Neuropediatrics 1986;17:33-8.
Dravet C, Bureau M. Benign myoclonic epilepsy in infancy. In: Roger J, Bureau M, Dravet Ch, Genton P, CA Tassinari, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey Ltd., 2002:69-79.**
Fusco L, Vigevano F. Ictal clinical and electroencephalographic findings of spasms in West syndrome. Epilepsia 1993;34:671-8.
Giovanardi Rossi P, Parmeggiani A, Posar A, Santi A, Santucci M. Benign myoclonic epilepsy: long-term follow-up of 11 new cases. Brain Dev 1997;19:473-9 **
Guerrini R, Dravet CH, Gobbi G, Ricci S, Dulac O. Idiopathic generalized epilepsies with myoclonus in infancy and childhood. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalized epilepsies: clinical, experimental, and genetic aspects. London: John Libbey Eurotext Ltd., 1994:267-80. **
Lin YP, Itomi K, Takada H, et al. Benign myoclonic epilepsy in infants: video-EEG features and long-term follow-up. Neuropediatrics 1998;29:268-71.
Loiseau P, Duche B, Loiseau J. Classification of epilepsies and epileptic syndromes in two different samples of patients. Epilepsia 1991;32:303-9.
Lombroso CT, Fejerman N. Benign myoclonus of early infancy. Ann Neurol 1977;1:138-43.
Pachatz C, Fusco L, Vigevano F. Benign myoclonus of early infancy. Epil Disord 1999;1:57-61.
Ricci S, Cusmai R, Fusco L, Cilio R, Vigevano F. Reflex myoclonic epilepsy of the first year of life. Epilepsia 1995;35:47.**
Todt H, Muller D. The therapy of benign myoclonic epilepsy in infants. In: Degen R, Dreifuss FE, editors. Epilepsy research. Suppl 6. The benign localized and generalized epilepsies in early childhood. Amsterdam: Elsevier, 1992:137-9.
Vigevano F, Cusmai R, Ricci S, Watanabe K. Benign epilepsies of infancy. In: Engel Jr J, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven Publishers, 1997:2267-76.
Детская миоклоническая эпилепсия характеризуется короткими по продолжительности приступами, представляющими собой кратковременные, часто симметричные мышечные сокращения в сочетании с резким снижением тонуса мышц туловища и внезапным падением вперед, нередко служащим причиной травм лица и повреждения полости рта. Миоклоническая эпилепсия представляют собой гетерогенную группу заболеваний с различной этиологией и вариабельным прогнозом. Тем не менее можно выделить по крайней мере пять различных подгрупп, охватывающих широкий спектр миоклонической эпилепсии в детской популяции.
Доброкачественный миоклонус младенчества дебютирует в младенческом возрасте и проявляется в виде серий миоклонических приступов, вовлекающих мышцы шеи, туловища и конечностей. По внешним проявлениям миоклонус можно ошибочно принять за младенческие спазмы, однако на ЭЭГ у пациентов с доброкачественным миоклонусом младенчества нет отклонений от нормы. Прогноз благоприятный, психомоторное развитие детей соответствует возрасту, миоклония прекращается к 2-летнему возрасту. Антиэпилептическая терапия не показана.
Развитие детей до дебюта миоклонических приступов детского возраста соответствует возрастной норме. Течение беременности и родов у матери ребенка без особенностей. Раннее развитие без патологии. Средний возраст ребенка в дебюте приступов — около 2 лет, однако возможен дебют приступов у детей от 6 мес. до 4 лет. Частота миоклонических приступов широко варьирует: у одних пациентов приступы возникают ежедневно, несколько раз в день, у других детей отмечаются межприступные периоды продолжительностью до нескольких недель.
У небольшой части пациентов дебюту миоклонической эпилепсии предшествуют фебрильные судороги или генерализованные тонико-клонические афебрильные приступы. Примерно в 50 % случаев у детей с миоклоническими приступами периодически возникают тонико-клонические приступы. На ЭЭГ быстрые комплексы пик-волна с частотой 2,5 Гц или выше в сочетании с нормальной основной активностью фоновой записи в большинстве случаев. По крайней мере в 1/3 случаев имеется эпилепсия в семейном анамнезе, что позволяет предположить наследственную этиологию заболевания.
Долговременный прогноз относительно благоприятный. Умственная отсталость отмечается в небольшом проценте случаев, и более чем у 50 % пациентов через несколько лет наступает ремиссия. Однако у значительной части детей наблюдаются речевые нарушения, трудности с обучением, эмоциональные и поведенческие расстройства, что требует длительного катамнестического наблюдения и адаптации детей с привлечением специалистов различных направлений.
Сложная миоклоническая эпилепсия это гетерогенная группа заболеваний с плохим прогнозом. Дебют парциальных или генерализованных тонико-клонических приступов наблюдается на первом году жизни и в типичных случаях предшествует дебюту миоклонической эпилепсии. Генерализованные судороги часто возникают на фоне инфекционного заболевания с поражением верхних дыхательных путей и субфебрильной лихорадкой; часто отмечается эпилептический статус. Примерно в Уз случаев диагностируется задержка психического развития. В анамнезе детей часто встречается указание на гипоксически-ишемическую энцефалопатию в перинатальном периоде; при определении неврологического статуса характерны признаки поражения пирамидных путей, экстрапирамидные расстройства в сочетании с микроцефалией. Указание на эпилепсию в семейном анамнезе реже встречается в этой группе пациентов по сравнению с детьми с типичной миоклонической эпилепсией.
У некоторых детей выявляется комбинация частых миоклонических и тонических приступов, в этих случаях при обнаружении на ЭЭГ медленных комплексов пик-волна заболевание диагностируется как синдром Леннокса-Гасто. Этот синдром характеризуется триадой резистентных к терапии приступов, медленной пик-волновой активностью на ЭЭГ в периоде бодрствования и умственной отсталостью. У пациентов со сложной миоклонической эпилепсией на межприступной ЭЭГ выявляются медленные пик-волновые разряды и резистентность к антиэпилептической терапии.
Приступы отличаются стойким характером, частота умственной отсталости и поведенческих нарушений достигает практически 75 % среди всех пациентов. Назначение препаратов вальпроевой кислоты или бензодиазепиновых производных может привести к снижению частоты или уменьшению выраженности приступов. У пациентов с приступами, резистентными к антиэпилептической терапии, следует рассматривать вопрос о назначении кетогенной диеты.
Доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ) – зависящая от возраста форма идиопатической эпилепсии, которая характеризуется генерализованными миоклоническими припадками. Этиология детально не изучена. Патология проявляется мышечными сокращениями верхних конечностей, шеи и головы длительностью в 1-3 сек. с частотою 2-3 раза в день. Общее состояние ребенка и его психофизическое развитие нарушается редко. Диагностика направлена на определение спайк- или полиспайк-волн на ЭЭГ. Основное лечение – медикаментозная монотерапия. Препараты выбора – вальпроаты, при их неэффективности используются бензодиазепины или производные сукцинимида.
- Причины ДМЭМ
- Симптомы ДМЭМ
- Диагностика ДМЭМ
- Лечение ДМЭМ
- Прогноз и профилактика ДМЭМ
- Цены на лечение
Общие сведения
Доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ) – редкая форма эпилепсии в педиатрии. Характерна только для определенной возрастной категории. Впервые заболевание выделено как отдельная нозологическая форма в 1981 году Дарве и Биором. Патология составляет менее 1% от всех форм эпилепсий и порядка 2% от ее идиопатических генерализованных форм. На данный момент в литературе описано около 100-130 случаев данного заболевания. ДМЭМ наблюдается у детей от 6 месяцев до 3 лет, в редких случаях возникает в возрасте до 5 лет. Представители мужского пола болеют в 1,5-2 раза чаще. Патология, как правило, хорошо поддается лечению и полностью купируется в старшем возрасте (в основном – после 6 лет). Осложнения в виде отставания в психомоторном развитии возникают редко и только при отсутствии терапии.
Причины ДМЭМ
Доброкачественная миоклоническая эпилепсия младенчества относится к числу генетически детерминированных заболеваний, передающихся по полигенному типу наследования. Является малоисследованной патологией, поскольку встречается довольно редко. ДМЭМ входит в группу идиопатических генерализованных эпилепсий, однако связи с другими нозологиями из этой группы не установлено. На данный момент неизвестно, мутация каких генов приводит к развитию ДМЭМ.
При сборе семейного анамнеза выясняется, что родители 40% больных страдают или страдали эпилепсией либо фебрильными припадками. Патогенетически развитие миоклонических атак обусловлено возникновением разряда быстрых генерализованных спайк-волн (СВ) или полиспайк-волн (ПСВ). Их частота составляет 3 Гц или более, а длительность – 1-3 сек. Волны формируются в лобных или теменных участках коры головного мозга. Сами атаки могут быть спонтанными или возникающими на фоне определенных (звуковых, тактильных или ритмичных световых) раздражителей.
Симптомы ДМЭМ
ДМЭМ диагностируется в возрасте от 6 месяцев до 3 лет. Развитие ребенка до появления первых миоклонических припадков проходит нормально. Примерно у 20% детей проявляются редкие судороги при рождении или в неонатальном периоде. Общее состояние пациента страдает редко, нарушения неврологического статуса не выявляются. Первые миоклонические атаки поражают верхние конечности, шею и голову, редко – ноги. Они могут иметь разную интенсивность, в том числе – у одного и того же ребенка во время разных эпизодов. Выраженность колеблется от еле заметных подергиваний до видимых фибрилляций.
Частота припадков составляет 2-3 раза в сутки с разными временными интервалами. Длинных серий атак не наблюдается. Возможна провокация атаки громким звуком, тактильной или ритмичной световой стимуляцией. После каждого эпизода может наблюдаться рефрактерный период длительностью от 20 до 120 сек. В этом временном промежутке даже интенсивная стимуляция не вызывает новый приступ. При этом часто наблюдается мышечная атония. Для заболевания характерно усиление миоклонических приступов при засыпании (дремоте) и их исчезновение в фазе медленного сна.
При тяжелых формах возможна генерализация судорог, сопровождающаяся потерей равновесия, внезапным выпадением предметов из рук, редко – расстройствами сознания. В процесс иногда вовлекаются межреберные мышцы, передняя брюшная стенка и диафрагма, из-за чего нарушается дыхание и может выслушиваться экспираторный шум. Для ДМЭМ характерно увеличение интенсивности клинических проявлений до определенного возраста и их последующее полное исчезновение. При длительном течении заболевания возможно отставание в психомоторном развитии. Трансформация в другие формы приступов, в том числе в абсансы, не происходит даже на фоне отсутствия специфического лечения.
Диагностика ДМЭМ
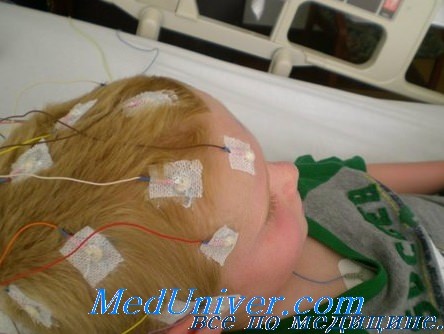
Диагностика доброкачественной миоклонической эпилепсии младенчества заключается в сборе анамнестических данных и проведении инструментальных методов исследования. Физикальное обследование ребенка в интериктальный период неинформативно. Лабораторные тесты каких-либо отклонений от возрастной нормы не выявляют. Наибольшую диагностическую ценность имеет повторная полиграфическая видеоэлектроэнцефалография (видео-ЭЭГ), которая позволяет обнаружить спайк-волны и доказать наличие миоклонических приступов. При необходимости проводится провокационная проба с ритмической световой или тактильной стимуляцией.
Вне приступов (а изредка – во время них) данные ЭЭГ остаются в пределах нормы, спонтанные спайк-волны возникают редко. Во время медленного сна возможно усиление разрядов в коре головного мозга при сохранении их нормальной структуры, возникновение быстрых ритмов или формальных изменений. В быструю фазу (REM-сон) могут фиксироваться генерализованные разряды спайк-волн. С целью исключения органической патологии могут назначаться нейросонография, компьютерная и магнитно-резонансная томография. При наличии клинической симптоматики на протяжении длительного периода проводится оценка психомоторного развития.
Дифференциальная диагностика ДМЭМ осуществляется с криптогенными детскими судорогами, доброкачественным неэпилептическим миоклонусом, синдромом Леннокса-Гасто и миоклонически-астатической эпилепсией раннего детского возраста.
Лечение ДМЭМ
Лечение ДМЭМ, как правило, проводится в амбулаторных условиях, исключением являются частые и тяжелые миоклонические атаки, требующие постоянного наблюдения. Показана медикаментозная терапия при помощи противоэпилептических средств. Первую линию составляют препараты из группы вальпроатов (натрия вальпроат). Важную роль играет подержание стабильной концентрации действующего вещества в крови. Нерегулярное введение назначенных средств провоцирует новые приступы и формирование резистентности к дальнейшей терапии данным медикаментом. При недостаточной эффективности вальпроатов показаны препараты из группы бензодиазепинов (нитразепам) или производных сукцинимида (этосуксимид). Терапевтический курс предполагает лечение на протяжении 3-4 лет с момента возникновения первых припадков.
При выраженной чувствительности к ритмичным световым раздражителям длительность курса увеличивается. При минимальной активности приступов или их исключительно рефлекторном характере лечение может проводиться в сокращенные сроки или не назначаться вовсе. При рецидиве миоклонических атак в старшем возрасте после пройденного лечения рекомендован упрощенный вариант аналогичного терапевтического курса. Обязательным моментом является психологическая поддержка семьи, непосредственно влияющая на эффективность лечения и направленная на исключение триггеров для ребенка.
Специфической профилактики для доброкачественной миоклонической эпилепсии младенчества не разработано. Прогноз в большинстве случаев благоприятный, заболевание, как правило, заканчивается полным выздоровлением ребенка. Миоклонические атаки, возникающие на фоне звуковой или тактильной стимуляции, прогностически более благоприятны, чем спонтанные. Переход в другие формы эпилепсии нехарактерен. Острый период, при котором наблюдаются выраженные припадки, в среднем длится менее 12 месяцев. Более чем у 53% детей в возрасте 6 лет все симптомы ДМЭМ полностью исчезают. Примерно у 14% в дальнейшем наблюдаются задержка психического развития или нарушения в поведении, из-за чего пациенты вынуждены получать образование в специализированных учебных заведениях. Частота осложнений напрямую зависит от своевременности диагностики, эффективности проводимого лечения и психологического климата в семье, в первую очередь – взаимоотношений между ребенком и матерью.
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ
Читайте также:
