
Эпилепсия идиопатическая парциальная с височной
Эпилепсия является весьма распространенным и хорошо изученным заболеванием, благодаря чему во многих случаях специалисты могут помочь полностью от него избавиться. Обращаясь к статистике, можно отметить, что в настоящее время около 1% всех жителей нашей планеты страдают этим заболеванием. В результате всевозможных исследований и наблюдений медицина выявила несколько форм эпилепсии, которые отличаются как причинами, по которым развивается болезнь и происходят приступы, так и по расположению её очага в головном мозге. В этой статье мы предлагаем вам ознакомиться с симптомами, причинами развития и методами лечения некоторых наиболее часто встречающихся типов заболевания.
Идиопатическая эпилепсия
Данная форма заболевания развивается не из-за органического поражения головного мозга человека. Идиопатическая эпилепсия возникает в результате изменения работы, выполняемой нейронами, когда они становятся более активными и увеличивается уровень их возбудимости.
Основные причины, по которым у человека может развиться идиопатическая эпилепсия:
Врожденные аномалии мозга
Отравление алкоголем или лекарственными препаратами
Одним из видов идиопатической эпилепсии является так называемая роландическая эпилепсия. Она встречается у детей и подростков в возрасте от трех до тринадцати лет. Свое название данный недуг получил из-за локализации его очага – он находится в роландовой борозде коры мозга.
Когда наступает приступ при роландической эпилепсии, то судороги происходят в мышцах лица и горла. Иногда могут происходить подергивания одной из конечностей.
Основные симптомы идиопатической роландической эпилепсии следующие:
Онемение и покалывание в области языка, губ
Судороги одной руки или ноги
Выделение слюны в больших количествах
Ухудшается память после приступа
Приступы происходят чаще ночью
Данная форма эпилепсии является доброкачественной, и она самостоятельно проходит примерно к шестнадцати годам. В большинстве случаев лечения не требует.
Идиопатическая эпилепсия является разновидностью фокальной эпилепсии.
Фокальная эпилепсия
Форма эпилепсии, называемая фокальной, даёт о себе знать в детском возрасте.
Причины развития фокальной эпилепсии могут быть разными.
Сотрясение мозга, сильный удар в область головы
Инфекции, имеющие вирусную природу
Воспаление тканей мозга
Травма, полученная при рождении
Симптомы фокальной эпилепсии зависят от того, в какой области головного мозга находится поражение.
Во время приступа у больного могут наблюдаться помутнения сознания, появляться навязчивые идеи, даже галлюцинации. Если приступ случается у ребенка, то его может мучить рвота, он сильно потеет и все время плачет. При поражении у больного только одной стороны мозга, может наблюдаться непроизвольное вращение белками глаз, подергивание пальцев на руках, судороги верхних конечностей.
В лечении фокальной эпилепсии используются медикаменты и различные процедуры, за исключением оперативного вмешательства. Также, при данном заболевании необходимо вести здоровый образ жизни, особе внимание стоит уделять качеству питания. Рекомендуется чаще есть продукты, которые способствуют восстановлению функционирования головного мозга. В основном это белки – куриное мясо, орехи, морская рыба и т.д. Будет весьма полезно отказаться от употребления кофе и спиртных напитков.
Височная эпилепсия
В большинстве случаев основной причиной данной формы эпилепсии является поражение височной доли при травмах, полученных во время родов. Но могут быть и другие причины. Например, совсем нередки случаи, когда данное заболевание развивалось у людей, перенесших черепно-мозговые травмы, такие инфекции как бруцеллез, клещевой энцефалит, гнойный менингит и другие. Кроме того, височной эпилепсией страдает немало людей, перенесших инсульт – как геморрагический, так и ишемический.
Когда у больного случается приступ, он может ощущать боли в животе, его может тошнить, а также нередко бывают боли в области сердца, одышка. Наблюдаются изменения сознания - панические атаки, истерика, человек теряет ориентацию в пространстве. Он может совершать действия, которые не имеют смысла в данный момент – например, снимать с себя одежду, брать разные предметы в руки и ставить их на другое место и т.п. Нарушается зрение, больной плохо воспринимает запахи.
Височная эпилепсия опасна тем, что все время прогрессирует. С течением времени у больных данной формой заболевания развиваются вегетативные расстройства, между приступами отмечается повышенная потливость, проблемы в эндокринной системе, аллергия.
Диагностика височной эпилепсии выполняется на основе показаний МРТ, ПЭТ, полисомнографии. Естественно, в первую очередь во внимание принимаются жалобы больного, его неврологический статус.
То, как именно будет производиться лечение височной эпилепсии, зависит от основной причины развития заболевания. Например, если заболевание вызвано инфекцией, то больному прописывают препараты, снимающие воспаление, а если причина болезни кроется в наличии опухолей в головном мозге, то пациента направляют на операцию. Но какой бы ни была причина развития височной эпилепсии, все равно больному показано противосудорожное лечение.
Профилактика данной формы заболевания заключается в своевременном лечении различных патологий головного мозга, предупреждении инфекций нервной системы, недопущение родовых травм головы.
Парциальная эпилепсия
Парциальная эпилепсия – это заболевание с хронической формой течения, оно развивается по причине повреждений нервных клеток, а также и из-за их высокой активности в одной части головного мозга.
Данная форма эпилепсии подразделяется на несколько подвидов:
Мультифокальная форма парциальной эпилепсии – это когда сразу несколько участков головного мозга оказываются пораженными.
Припадки парциальной эпилепсии бывают трех видов:
Простые (когда отсутствуют нарушения сознания)
Сложные (когда имеют место быть нарушения сознания)
Вторичной генерализации (когда в процесс вовлекается все тело больного)
Когда речь идёт о симптомах данной формы эпилепсии, то обязательно учитывается место локализации очага в одном из участков мозга. К примеру, если поражена лобная часть, то приступы в большинстве случаев случаются по ночам, их длительность заключается между 30 и 60 секундами. Могут быть судороги верхних или нижних конечностей, больной начинает часто моргать, поворачивать голову, вращать белками глаз, его лицо краснеет, а речь становится прерывистой.
В случае с поражением головного мозга в области виска, у больного происходят сложные приступы, когда он теряет сознание или застывает, широко раскрыв глаза.
Теменная форма парциальной эпилепсии даёт о себе знать через покалывание или ощущение жжения, онемением рук или ног, болью в разных частях тела.
При затылочной эпилепсии у больного нарушается зрение, могут наблюдаться зрительные галлюцинации, непроизвольные повороты головы.
Диагностируется данная форма заболевания при помощи комплексного обследования у невролога, а также прохождения больным электроэнцефалографии, МРТ, компьютерной томографии, проводится также и исследование глазного дна. Нередко людей, у которых подозревают наличие парциальной эпилепсии, отправляют на консультацию к психиатру.
Лечится данная форма эпилепсии медикаментозно, в основном, назначаются противосудорожные препараты. Если же такой метод лечения не даёт положительных результатов, то больного направляют в отделение нейрохирургии, где он проходит соответствующее лечение.
Профилактика эпилепсии заключается, в первую очередь, в ведении здорового образа жизни. Для того, чтобы не допустить припадочных приступов, необходимо отказаться от курения, перестать употреблять спиртные напитки, в том числе и те, которые относятся к легким. Кроме того, нужно уменьшить потребление чая и кофе, в особенности крепкого. Не рекомендуется есть перед сном, подниматься на высоту. Врачи советуют больным парциальной эпилепсией чаще гулять на свежем воздухе и потреблять кисломолочные продукты.
Джексоновская эпилепсия

Это форма эпилепсии отличается тем, что приступы у больных происходят специфически – на той или иной (но только одной) части тела. Свое название данное заболевание получило благодаря ученому, невропатологу из Англии по фамилии Джексон – именно он открыл эту форму эпилепсии.
Особенностью джексоновской эпилепсии является тот факт, что приступы с больными случаются при полном сознании. Приступ берет свое начало в одной из конечностей и распространяется дальше, но только по этой же стороне тела, не переходя на другую. Чаще всего все начинается с пальцев руки, потом приступ доходит до плеча, затрагивает лицо, перекидывается на ногу. Проходит приступ в обратном порядке.
Причины, по которым чаще всего у человека может развиваться джексоновская эпилепсия, следующие:
Травмы головы и, как следствие, поражения головного мозга
Сосудистые аномалии мозга
Опухоли головного мозга
Лечение джексоновской эпилепсии осуществляется медикаментозно, с применением противосудорожных средств. Но если медикаментозная терапия не даёт никакого результата, то тогда прибегают к оперативному вмешательству.
Юношеская миоклоническая эпилепсия
Данная форма является одной из самых распространенных. Её часто называют ещё и синдромом Янца, а также миоклонус-эпилепсией. Чаще всего она развивается в возрасте от 8 до 26 лет, но первые признаки в большинстве случаев наблюдаются в возрасте между 12 и 16 годами, преимущественно у мальчиков. Известны случаи, когда миоклоническая эпилепсия развивалась у младенцев.
Причины развития миоклонической эпилепсии:
Повреждения мозга в результате травмы головы
Повреждение мозга ребенка во время беременности
Новообразования в головном мозге
Нарушение кровообращения в мозге
Нередко врачам так и не удается установить реальную причину возникновения заболевания. Это относится к некоторым частным случаям в медицинской практике.
Главным признаком, свидетельствующим о наличии у человека такого заболевания, как миоклоническая эпилепсия, является эпилептический припадок.
Приступы у страдающих данным недугом могут быть трех типов:
Миоклонические : характеризуются резким, неожиданным подергиванием мышц, конечностей, лица или всего тела. Часто это случается сразу после пробуждения, порою во время утреннего приема пищи. Если больной сильно устает в течение дня, то приступ у него может случиться и в вечерние часы.
Тонико-клонические : они случаются у 60 процентов пациентов, страдающих юношеской миоклонической эпилепсией. Они бывают в первые часы после пробуждения. Чаще они происходят из-за того, что больной слишком поздно лег спать или слишком рано проснулся.
Абсансы : наблюдаются у одной трети или даже половины больных юношеской миоклонической эпилепсией. Во время таких приступов человек теряет сознание примерно на полминуты. Судорог, как правило, нет. Чаще всего абсансы бывают утром, но, в принципе, случаются и в любое другое время суток.
Приступ при юношеской миоклонической эпилепсии может случиться и после употребления спиртного.
Лечение данной формы заболевания нервной системы осуществляется посредством противоэпилептических препаратов. За счет них удается снизить длительность и количество припадков, а в некоторых случаях и вовсе избавиться от них. Для того, чтобы достичь положительного результата, необходимо принимать препараты регулярно и в течение длительного времени. Часто больным приходится принимать лекарства всю жизнь.
Проявления приступов эпилепсии могут отличаться у разных больных. В первую очередь, они зависят от тех зон головного мозга, где возникает и распространяется патологический разряд. В этом случае симптомы будут напрямую связаны с функциями данных отделов. Могут происходить двигательные расстройства.
Эпилепсия – это заболевание хронического характера, поражающее головной мозг больного и сопровождающееся предрасположенностью к судорогам с отключением сознания. Однако, несмотря на хроническое течение патологии, её лечение возможно, оно может быть медикаментозным и хирургическим.
Эпилепсия – болезнь неизлечимая и относится к разряду хронических. Поэтому, хотя и существует множество способов оказания помощи больным людям, тем не менее, приступ может случиться в любой момент. В прошлом эпилептический припадок вводил людей в.
Нельзя держать кухонную утварь из керамики и стекла, её безопасной альтернативой является пластиковая посуда. Колбасные изделия, сыр, хлеб и другие продукты покупаются нарезанными. Желательно реже пользоваться ножами, держа их в закрытом месте. От газовой плиты лучше.
Чтобы добиться хороших результатов, лекарственные растения необходимо использовать в лечении эпилепсии довольно длительное время. Нередко настои из них приходится пить в течение нескольких лет. При этом рекомендуется не употреблять все время только одно растение, а.
Длительное ограничение в питательных веществах, поступаемых в организм из различных продуктов, позволяет значительно снизить частоту эпилептических припадков. В некоторых случаях приступы заболевания полностью исчезают, и пациенты могут вести полноценный образ жизни.
Как и при подавляющем большинстве заболеваний, при эпилепсии имеется некоторое количество групп инвалидности: от первой до третьей. В соответствии с международной статистикой, их различают между собой по следующим критериям, которые обязательно следует знать тем, кто претендует на.

Эпилепсии известны с давних времен, еще с глубокой древности. Их интенсивное изучение проводилось на протяжении предыдущего столетия, особенно в последние десятилетия. За это время знания углублялись, а взгляды на эпилепсии как заболевания менялись, прошли несколько этапов.
На первом этапе считали, что эпилепсии – это наследственные заболевания. В США существовали законы (только в 1982 г. был отменен последний), согласно которым больным эпилепсией запрещалось вступать в брак и иметь детей. Такие больные подвергались принудительной стерилизации.
Второй этап был связан с бурным развитием методов диагностики, использованием новых достижений науки и техники. Этот период характеризовался полным отрицанием роли генетических факторов в возникновении эпилепсий. После появления методов нейровизуализации и их широкого применения в обследовании больных эпилепсиями утвердилось мнение, что все эпилепсии являются симптоматическими. Исследователи стали считать, что в основе всех случаев эпилепсий лежат органические поражения головного мозга, если же эти поражения выявить не удается, то это связано с тем, что методы диагностики недостаточно совершенны, в дальнейшем, с появлением новых, более совершенных, причина возникновения эпилепсий (этиология) всегда будет установлена.
На современном этапе признается существование как симптоматических, так и генетически обусловленных эпилепсий. Существует мнение, что влияние на манифестацию эпилепсий оказывают 2 фактора: наследственная отягощенность и внешние воздействия (инфекции, травмы, интоксикации и др.).
На схеме показано влияние различных факторов на возникновение эпилепсий (В.А. Карлов, 2001).
За последнее десятилетие удалось картировать гены многих эпилептических синдромов, которые являются составляющей таких наследственных болезней, как болезнь Лафора, сиалидоз, болезнь Гоше, галактосиалидоз и др. (табл. 1).
Кроме заболеваний с картированными генами, при которых эпилептический припадок является одним из синдромов в структуре заболевания, существуют идиопатические эпилепсии (ИЭ), при которых эпилептический припадок является основным и, в большинстве случаев, единственным признаком заболевания.
При некоторых формах эпилепсий полный набор генов, с которыми ассоциируется каждое из заболеваний, не установлен. К таким заболеваниям относят доброкачественную эпилепсию детского возраста с затылочными пароксизмами, доброкачественную парциальную эпилепсию (ПЭ) с аффективными пароксизмами, семейную височную эпилепсию и первичную эпилепсию чтения.
При некоторых формах эпилепсий известно только то, что за возникновение и развитие болезни отвечает один или несколько генов. Моногенные и полигенные эпилепсии представлены в таблице 2.
К формам эпилепсий с установленными генами относят: доброкачественные семейные припадки новорожденных, генерализованные эпилепсии с фебрильными судорогами (+), аутосомно-доминантную ночную лобнодолевую эпилепсию и аутосомно-доминантную ПЭ со слуховыми симптомами (табл. 3).
Ранее считали, что ИЭ могут быть только генерализованные формы. Однако в процессе исследований было установлено, что некоторые ПЭ также являются идиопатическими. К настоящему времени установлены следующие идиопатические парциальные эпилепсии:
- Роландическая или доброкачественная эпилепсия (РЭ) детского возраста с центротемпоральными пиками.
- Доброкачественная ПЭ детского возраста с затылочными пароксизмами (синдром Гасто) и ее вариант, отмечающийся у детей более раннего возраста – синдром Панайотопулоса.
- Доброкачественная ПЭ с аффективными симптомами (синдром Далла-Бернардина).
- Аутосомно-доминантная лобная эпилепсия с ночными пароксизмами.
- Семейная височная эпилепсия.
- Эпилепсия чтения.
- ПЭ со слуховыми симптомами;
- Семейная ПЭ с вариабельным фокусом.
Только 3 из всех идиопатических парциальных эпилепсий дебютируют исключительно в детском возрасте. К таким формам относят РЭ, синдромы Гасто и Панайотопулоса, синдром Далла-Бернардина.
ИЭ определяются по следующим признакам:
- возрастзависимый дебют;
- наличие припадков только одного типа, отсутствие их трансформации;
- отсутствие неврологической симптоматики;
- отсутствие интеллектуально-мнестических и психических нарушений;
- эффективность медикаментозного лечения, в большинстве случаев – монотерапии;
- благоприятный прогноз в виде полного исчезновения приступов (в большинстве случаев).
Основные различия идиопатических и симптоматических эпилепсий представлены в таблице 4.
Роландическая эпилепсия
Из всех форм идиопатических парциальных эпилепсий в детском возрасте зачастую встречается РЭ (доброкачественная ПЭ с центрально-темпоральными пиками). Доля этой формы эпилепсий составляет 10-20% от всех эпилепсий детского возраста.
Дебют РЭ наблюдается в возрасте 2-12 лет (пики возникновения заболевания приходятся на возраст 3 и 9 лет). Мальчики болеют чаще, чем девочки. Клиническая характеристика приступов заключается в следующем:
- возникают в ночное время суток;
- короткие (длительность до нескольких минут);
- после пробуждения или в дневное время суток ощущаются, предшествующие приступу, парестезии в области языка, глотки;
- наблюдаются гемифациальные или фацио-брахиальные подергивания, сопровождающиеся выраженным слюнотечением;
- припадки иногда генерализуются, захватывая все тело.
После окончания припадка временно пропадает речь. Припадки редкие, с частотой 1-2 в месяц.
Полная терапевтическая ремиссия наступает до возраста 15 лет в 97% случаев.
Базовыми препаратами для лечения РЭ являются препараты вальпроевой кислоты в суточной дозе 20-30 мг/кг массы тела. В случаях их неэффективности рекомендуются карбамазепины (10-20 мг/кг/сут) или дифенин (3-5 мг/кг/сут). Политерапия и применение барбитуратов противопоказаны.
Синдром Панайотопулоса
Для синдрома Панайотопулоса характерен возраст дебюта 1-9 лет, пик возникновения заболевания приходится на 3-6 лет. Типичным является возникновение припадков в ночное время. Приступы очень длительные, от 30 минут до 7 часов (в среднем 2 часа). Клиническими особенностями припадков являются вегетативные проявления, длительная утрата сознания, тенденция к статусному течению.
При синдроме Панайотопулоса в 93% эпилептические припадки сопровождаются вегетативными проявлениями. Из всех вегетативных симптомов наиболее частыми являются тошнота, позывы на рвоту, рвота. Кожные покровы во время припадка бледные, реже отмечается их покраснение; зрачки чаще всего расширены; нарушение сознания сопровождается недержанием мочи и кала. Практически всегда наблюдаются слабо выраженные нарушения дыхания и сердечного ритма, но в некоторых случаях достигающие остановки сердца. Иногда отмечается гиперсаливация, нарушение моторики кишечника, нарушение терморегуляции.
Сознание нарушено у 94% больных. Практически у всех пациентов наблюдается отведение глазных яблок в одну из сторон. Во время приступа ребенок обмякает, реже (в 26%) наступают гемиконвульсии, еще реже (в 20%) – генерализованные тонико-клонические или клонико-тонические судороги.
У 44% детей с этим синдромом наблюдается так называемый вегетативный эпилептический статус, длительностью до 7 часов.
После эпилептического припадка наступает сон, длящийся несколько часов. По его окончании каких-либо субъективных или объективных нарушений не отмечается.
Частота припадков невелика, иногда за все время заболевания бывает только 1 припадок.
Использование компьютерной томографии или магнитно-резонансной томографии не выявляет поражений мозговых структур. Дети имеют нормальное психоэмоциональное и интеллектуальное развитие.
При ЭЭГ-исследовании фиксируется медленная активность со спайками (у 2/3 пациентов только в затылочных отведениях). ЭЭГ-нарушения проявляются только при записи с закрытыми глазами. Если пациент открывает глаза, изменения исчезают. ЭЭГ пациента с синдромом Панайотопулоса представлена на рисунке 3.
Ремиссия наступает к 9 годам у 92% пациентов.
Синдром Гасто
Эпилепсия Гасто характеризуется более поздним началом – от 3 до 15 лет, средний возраст дебюта заболевания – 8 лет.
Приступы чаще проявляются в дневное время суток (2/3 случаев), бывают очень короткими (от нескольких секунд до 3 минут), реже отмечаются при пробуждении.
Клиническая картина: наличие парциальных сенсорных приступов со зрительными нарушениями, элементарных зрительных галлюцинаций, частые случаи кратковременной слепоты или частичной потери зрения, реже встречаются сложные зрительные галлюцинации и иллюзии. Версивный компонент с поворотом головы и глаз контрлатерально очагу наблюдается у 70% больных. Также имеются симптомы распространения возбуждения из затылочной доли в виде гемиконвульсий (43%), комплексных фокальных припадков (14%), дисфазии, адверсивных припадков (25%), вторично-генерализованных тонико-клонических судорог (13%).
Постприступная мигреноподобная головная боль встречается у половины больных. Иктальная рвота бывает очень редко.
Прогноз: ремиссия в 82% к 15 годам.
В таблице 5 представлены различия между синдромами Панайотопулоса и Гасто.
Для предотвращения припадков назначается базовый препарат – вальпроат (30-50 мг/кг в 2-3 приема), а также препараты – карбамазепин (20-30 мг/кг/сут) и клоназепам (0,15 мг/кг/сут). При отсутствии адекватного лечения приступы могут возникать спонтанно, во время разговора и т.д.
Синдром Далла-Бернардина возникает в возрасте от 2 до 9 лет, клинически проявляется приступами внезапного страха или ужаса, сопровождающимися жевательными, глотательными автоматизмами, остановкой речи, вегетативными симптомами, гиперкинезами, абортивными болями. Приступы развиваются сразу после засыпания или в дневное время суток с продолжительностью в 1-2 минуты.
Препаратами 1-й линии выбора при синдроме Далла-Бернардина являются карбамазепины (10-20 мг/кг/сут массы). При их неэффективности могут применяться дифенин (2-3 мг/кг/сут) и препараты вальпроевой кислоты (20-30 мг/кг/сут). Показана исключительно монотерапия. В целом прогноз благоприятный, в редких случаях приступы могут сохраняться у пациентов старше 18 лет.
Результатом генетических исследований является выделение идиопатических парциальных эпилепсий в самостоятельные нозологические формы. Углубление знаний детских неврологов об особенностях клинической картины и течения, ЭЭГ-паттернов и результатах нейровизуализации, характерных для этих форм заболевания, позволяют своевременно отдифференцировать их от симптоматических парциальных эпилепсий. Это имеет принципиальное значение в выборе адекватной терапии – лечения с помощью монотерапии, назначения препаратов вальпроевой кислоты, наиболее эффективных при идиопатических формах. Идиопатические парциальные эпилепсии, по сравнению с симптоматическими, имеют более благоприятный прогноз, что определяет тактику ведения – отсутствие необходимости длительного лечения, возможность отмены антиэпилептических препаратов в определенные возрастные периоды.
В настоящее время эпилепсия входит в пятерку наиболее распространенных неврологических заболеваний. Она нередко дебютирует уже в детском возрасте и относится к потенциально инвалидизирующим патологиям. Причем в большинстве случаев диагностируется идиопатическая форма эпилепсии. Это классический и наиболее часто встречающийся вариант заболевания, с несколькими клиническими разновидностями и вариантами течения.
Что это за болезнь

Эпилепсией называют хроническое заболевание, обусловленное патологией на уровне головного мозга и проявляющееся преимущественно приступами (припадками) судорожного и бессудорожного характера. Свойственны ей и другие, менее яркие симптомы. Изучением всех связанных с эпилептической болезнью проблем занимается особая наука эпилептология, но в повседневной клинической практике диагностику и лечение нередко проводит невролог (невропатолог).
Согласно действующей классификации, эпилепсия подразделяется на идиопатическую, симптоматическую и криптогенную. Эти неоднородные по происхождению и симптоматике состояния отличаются друг от друга характером изменений в центральной нервной системе.
Идиопатическая эпилепсия относится к самостоятельно возникающим заболеваниям. Раньше ее называли истинной, генуинной, первичной. Она характеризуется несколькими признаками:
- В головном мозге нет органических (структурных) изменений. Поэтому такой форме болезни не свойственны очерченная очаговая неврологическая симптоматика, задержка психомоторного развития или когнитивный регресс. Современные высокоточные методы нейровизуализации оказываются не информативными.
- Приводящие к припадкам нарушения носят функциональный характер и не обусловлены действием каких-либо внешних факторов или появившихся в течение жизни заболеваний.
- Склонность к раннему проявлению симптоматики. Идиопатическая эпилепсия в 70-75% случаев дебютирует в детском возрасте, с основным пиком заболеваемости у подростков в 9-14 лет. Поэтому обычно речь идет о детских и ювенильных (юношеских) формах болезни.
- Наследственная предрасположенность. В настоящее время выделено около 500 генов в соматических хромосомах, мутация в которых может стать причиной эпилепсии. Многие формы болезни характеризуется аутосомно-доминантным или рецессивным типом наследования.
- Достаточно высокая чувствительность к современным противосудорожным препаратам. Это позволяет в большинстве случаев добиться значительного урежения приступов или даже перевести заболевание в стойкую ремиссию.
На долю идиопатических типов приходится около 70-75% случаев эпилепсии с различными клиническими проявлениями, при этом преобладают ее генерализованные формы.
Что происходит в мозге при идиопатической эпилепсии
Болезнь не связана с локальной гибелью или ишемией (выраженным кислородным голоданием) нервных клеток, опухолевым ростом, внутричерепной гипертензией, пороками развития или какими-либо другими патологическими процессами. Все изменения носят функциональный характер и формируются на клеточном и биохимическом уровнях.
Согласно современным представлениям, значительная часть случаев идиопатической эпилепсии обусловлена каналопатией – дисфункцией ионных каналов в стенках нервных клеток. Такая проблема обычно обусловлена патологией одного или нескольких генов, кодирующих структуру трансмембранных белков.
Результатом подобных каналопатий становится дефект транспорта отдельных ионов (калия, натрия) или целых молекул нейромедиаторов (ГАМК, ацетилхолина и др.), что приводит к комплексу нарушений:
- изменение скорости проведения сигналов в нервной ткани, склонность к каскадному распространению пароксизмальных импульсов;
- поддержание дисбаланса между процессами торможения и возбуждения в головном мозге, с преобладанием активирующих влияний;
- создание предпосылок для чрезмерной стойкой деполяризации на поверхности нейронов (она может быть запущена выраженным ионным дисбалансом или избыточным количеством глутамата, причем в подавляющем большинстве случаев без явного провоцирующего фактора).
При идиопатической эпилепсии соседние нейроны очень легко синхронизируются друг с другом, что объясняет склонность к быстрому тотальному распространению пароксизмально возникшего гипервозбуждения. В этот процесс генерализации вовлекаются и подкорковые регулирующие структуры, что ускоряет передачу эпилептического импульса. В результате возбуждение охватывает практически весь головной мозг, провоцируя приступ с различной симптоматикой.
При этом у каждого пациента формируется собственная достаточно стабильная схема вовлечения и реакции различных мозговых структур. Это определяет характерную клиническую картину болезни, с преобладанием определенных симптомов во время приступа.
Основные проявления
Ключевое проявление эпилепсии – разнообразные приступы, которые могут комбинироваться друг с другом или существовать в изолированном виде. Они бывают судорожными (сопровождающимися неконтролируемой пароксизмальной активностью скелетных мышц) и бессудорожными, фокальными и генерализованными.
Нередко развитию приступа предшествует аура. А при серии следующих друг за другом припадков с генерализацией говорят о развитии эпилептического статуса, что чревато развитием отека головного мозга.
Судорожные приступы
При идиопатической эпилепсии возможно появление приступов любого типа. Но преобладание одного из них или характерная трансформация разных видов друг в друга позволяет дифференцировать формы болезни.
К основным вариантам припадков относят:
Для большинства идиопатических форм болезни не свойственно развитие неврологического дефицита и выпадение высших корковых функций. Физическое и интеллектуальное развитие у детей обычно не страдает, если нет осложняющих ситуацию сопутствующих заболеваний и предшествующих перинатальных вредностей.
Изменения личности по эпилептическому типу возникают нечасто, лишь при тяжело протекающих формах болезни с частыми плохо контролируемыми генерализованными приступами и эпистатусами.
Классификация
В настоящее время общепризнанной считается классификация, основы которой были приняты еще в 1981 году Международной Лигой по борьбе с эпилепсией (Киото, Япония). При этом учитывается характер имеющихся у пациента приступов, с оценкой их разновидности и склонности к генерализации.
В соответствии с таким подходом, все варианты болезни подразделяются на 2 основные группы: генерализованные и парциальные (фокальные). Внутри каждой их них имеется несколько различных заболеваний, отличающихся по клинической картине, времени проявления симптомов и прогнозу.
Выделяют несколько основных клинических разновидностей (по действующей классификации ILAE от 1989 г.):
- Детская абсансная эпилепсия – бессудорожный вариант заболевания с доказанной наследственной природой. Характеризуется частыми многократными приступами по типу абсанса у детей 4-9 лет. На нее приходится 10-17% от всех идиопатических форм.
- Ювенильная (юношеская) абсансная эпилепсия, с дебютом в подростковом возрасте. Диагностируется примерно в 12% случаев, у ⅔ пациентов дебютирует в возрасте 9-13 лет. При этом первоначально у ребенка могут возникать и типичные генерализованные судорожные приступы, а в последующем преобладают абсансы.
- Доброкачественная младенческая миоклонус-эпилепсия. Дебютирует в возрасте от 4 мес. до 3 лет с миоклонических вздрагиваний, в последующем эти приступы приобретают генерализованный характер. Чаще заболевают мальчики.
- Ювенильная миоклонус-эпилепсия или синдром Янца. Распространенность колеблется от 4 до 12%. Патология связана с дефектом генов на коротком плече 6 соматической хромосомы. В клинике преобладают миоклонические припадки, хотя они могут дополняться эпизодически возникающими абсансами и генерализованными судорожными приступами.
- Идиопатическая эпилепсия с генерализованными судорожными приступами (изолированными).
- Эпилепсия с генерализованными судорожными приступами периода пробуждения. Заболевание с доказанной генетической детерминированностью и дебютом преимущественно в 9–11-летнем возрасте. Характеризуется появлением типичных генерализованных припадков с тонико-клоническими судорогами в просоночном состоянии.
- Эпилепсия с фотосенсибилизацией. Характеризуется рефлекторным возникновением приступов в ответ на зрительную стимуляцию, с преобладанием генерализованных судорожных тонико-клонических припадков. Пик заболеваемости приходится на возраст 12-14 лет, но первичные проявления возможны вплоть до 25-летнего возраста.
Генерализованная идиопатическая эпилепсия – самый частый вариант заболевания, именно она определяет почти ⅔ случаев эпиприпадков в детском и подростковом возрасте.
Эта полиморфная группа включает несколько заболеваний.
- Доброкачественные семейные судороги у новорожденных или доброкачественная семейная неонатальная эпилепсия
Один из прогностически благоприятных вариантов заболевания, несмотря на очень ранний дебют симптоматики. Первые проявления отмечаются обычно уже на 1-й неделе жизни ребенка, в виде фокальных коротких приступов с апноэ. В последующем могут присоединяться двигательные автоматизмы или клонические подергивания. Симптоматика обычно угасает к началу 2-го года жизни. Заболевание относится к редким и имеет аутосомно-доминантный тип наследования.
- Идиопатическая парциальная эпилепсия с лобными автоматизмами
Дает о себе знать обычно в возрасте 2-8 лет. Характерна дифференцировка симптоматики в ночное и дневное время. В состоянии бодрствования у ребенка преобладают сложные парциальные и абсансоподобные приступы, а во сне появляются гемифациальные (с вовлечением половины лица) моторные припадки, которые иногда имеют склонность к вторичной генерализации. Активный период болезни длится несколько лет, после чего обычно наступает стойкая ремиссия.
- Затылочная доброкачественная эпилепсия с поздним началом (синдром Гасто)
Пик заболеваемости в 8-9 лет. Проявляется непродолжительными сложными по структуре фокальными приступами: начало припадка со зрительных галлюцинаций, которые сменяются кратковременным исчезновением зрения (корковой слепотой) и односторонними клоническими судорогами. Выход из приступа через мигренозные боли.
- Семейная височная эпилепсия
Характеризуется простыми или сложными парциальными припадками и предшествующей психосенсорной аурой. Один из вариантов болезни – приступы в виде слуховых галлюцинаций, иногда дополняющиеся вегетативными расстройствами. Дебютирует обычно в подростковом возрасте, но нередко первые ее проявления возникают лишь к 25-30 годам. Очень хорошо поддается медикаментозной терапии и не склонна к трансформации или генерализации.
В последние годы некоторыми исследователями выделяется еще одна особая форма болезни: идиопатическая фокальная эпилепсия с псевдогенерализованными приступами. У пациентов с таким диагнозом изначально возникающие фокальные приступы быстро развертываются и усложняются, что клинически выглядит как их генерализация. Но при этом ЭЭГ однозначно показывает, что происходит лишь вторичная билатеральная синхронизация, без тотального охвата всего мозга.
К редким формам заболевания относится синдром Панайотопулоса или идиопатическая затылочная эпилепсия с ранним началом, дебютирующая в возрасте от 1 до 13 лет. Характеризуется тяжело протекающими приступами с ротацией (поворотом) глаз, выраженным вегетативным компонентом и длительно сохраняющимся бессознательным состоянием. Чаще всего они начинаются в период сна, и нередко первым проявлением припадка становится не приносящая облегчения рвота.
Риск развития эпилептического статуса при синдроме Панайотопулоса очень высокий. Но заболевание в целом оценивается как прогностически благоприятное, с доброкачественным течением и сохранностью у ребенка интеллектуально-мнестических функций. В большинстве случаев приступы возникают редко, всего 1-3 раза в течение жизни, не оказывают негативного влияния на развитие ребенка и на общее состояние его здоровья.
Диагностика
Обследование включает консультацию невролога, функциональную диагностику (ЭЭГ, сомнография, суточное мониторирование) и современные методы нейровизуализации для исключения органической причины припадков.
Чаще всего для исследования головного мозга назначают МРТ или КТ, предпочтительно с ангиопрограммой и дополнительным контрастным усилением. Это позволяет исключить объемные образования, пороки развития, выраженные нарушения ликвородинамики, сосудистые изменения (в том числе врожденные мальформации) и другую патологию, способную привести к очаговой эпилептической активности. Такое обследование необходимо для дифференциальной диагностики и подтверждения идиопатической природы заболевания.
Информативность ЭЭГ может быть различной. Это зависит от периода, когда проводилось исследование, формы и тяжести течения болезни. В приступный период ЭЭГ выявляет генерализованную синхронизированную билатеральную (двустороннюю, с охватом обоих полушарий головного мозга) эпилептическую активность по типу пик-волна. Разряды короткие, обычно провоцируются гипервентиляцией и фотостимуляцией, вероятность их выявления повышается при депривации (целенаправленном лишении) сна.
В большинстве случаев в межприступную (интериктальную) фазу ЭЭГ не выявляет полипиковой и генерализованной судорожной активности, общий (основной) ритм сохранен. Функциональные признаки структурных дефектов не характерны. Исключение составляет ювенильная миоклонус-эпилепсия, при которой аномальная ЭЭГ выявляется практически постоянно у 80-95% пациентов.
При некоторых формах заболевания могут выявляться также другие аномалии: доминирующий окципитальный (затылочный) дельта-ритм, эпизодическое бифронтальное (в лобных отделах обоих полушарий) замедление ритма и др.
Как лечить
Пациентам с любыми эпилептическими припадками показана диспансеризация с регулярным контролем состояния. Лечение подбирается индивидуально неврологом или эпилептологом. Иногда требуется также участие психиатра, если клиническая картина включает психовегетативные пароксизмы, обманы восприятия (галлюцинации), нарушения поведения и когнитивные расстройства.
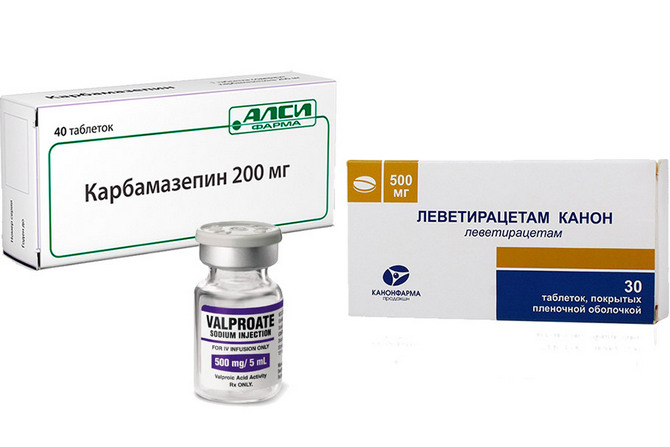
Противосудорожные препараты применяемые в терапии идиопатической эпилепсии
Основой лечения является медикаментозная терапия, с использованием антиконвульсантов (противосудорожных средств) различных групп. К наиболее употребимым препаратам относят:
- препараты вальпроевой кислоты (вальпроаты);
- Леветирацетам (Топирамат);
- Карбамазепин.
Препараты и схема их приема подбираются индивидуально, с учетом вида возникающих у пациента припадков. Лечение начинают с назначения малых доз средства 1 линии, с последующим подбором дозировки. При его недостаточной эффективности принимается решение о смене препарата или переходе на комбинированную терапию.
Противосудорожные средства принимаются постоянно, при этом необходимо регулярно контролировать картину крови и биохимические показатели работы печени. Продолжать лечение рекомендуется в течение не менее 5 лет после последнего приступа, отмена препаратов проводится постепенно, под контролем врача.
У 3-5% пациентов встречается медикаментозная резистентность – устойчивость к применяемым лекарствам, с отсутствием положительной динамики даже на комбинированной терапии достаточными дозами. В этом случае при некоторых формах заболевания может быть принято решение о целесообразности хирургического лечения, с проведением резективных или функциональных операций на головном мозге. Иногда проводят также стимуляцию блуждающего нерва.
К немедикаментозным методикам относят соблюдение режима дня, исключение провоцирующих (стимулирующих) факторов и диету. Желательно избегать потребления кофе, крепкого чая, чрезмерно острой пищи, алкоголя, тонизирующих напитков. Иногда врач дает рекомендации о переводе пациента на кетогенную диету – особую систему питания, когда ведущим источником энергии становятся жиры, а углеводы в рационе строго ограничиваются.
Прогноз
В целом идиопатические формы эпилепсии имеют относительно благоприятный прогноз. Многие из них дебютируют в детско-подростковом возрасте и в последующем постепенно угасают, иногда давая о себе знать лишь при явных провоцирующих факторах у взрослых.
Исключение составляют некоторые тяжело протекающие формы эпилепсии. Резистентность (устойчивость) к терапии нередко отмечается также у абсансных форм болезни, что требует тщательного подбора комбинации препаратов. Но даже в этом случае у ⅓ пациентов удается достичь лишь улучшения в виде значительного урежения и упрощения абсансов и исчезновения дополняющих их судорожных припадков.
Возможна также трансформация форм болезни друг в друга, несмотря на изначальное поражение разных генов. Например, эпилепсия с генерализованными приступами пробуждения может перейти в ювенильные формы абсансной или миоклонической эпилепсии. А у 30% девочек она в последующем трансформируется в катамениальную эпилепсию, при которой первично-генерализованные судорожные приступы возникают в предменструальный период.
Но в большинстве случаев удается добиться хорошего медикаментозного контроля над частотой и выраженностью приступов, избежать социально-бытовой дезадаптации пациентов и сохранить хороший темп интеллектуального развития ребенка. В стойкую ремиссию уходят 65-85% пациентов, причем большинству из них во взрослом возрасте уже не требуется постоянная поддерживающая терапия.
Читайте также:
