
Ранняя миоклоническая эпилептическая энцефалопатия младенцев
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ
Криптогенные и/или симптоматические
Таблица 3.8. Сравнительная характеристика инфантильных энцефалопатий
Обозначения: + наличие признака; — отсутствие признака.
Характеризуя ранние возрастзависимые эпилептические энцефалопатии, можно выделить несколько общих особенностей:
• жесткая зависимость от возраста; невозможность возникновения конкретной эпилептической энцефалопатий вне определенного возрастного диапазона;
• тяжелые и продолженные во времени эпилептические изменения на ЭЭГ;
• выраженная гетерогенность этиологии;
• частая ассоциация с моторными и ментальными нарушениями;
• весьма значительные затруднения в лечении и относительно неблагоприятный прогноз.
Такое значительное сходство указанных синдромов, а также использование нейроонтогенетического подхода при анализе ранних детских эпилепсий позволяют предположить, что именно ранний возраст, т.е. структурная и функциональная незрелость головного мозга, является основным фактором, детерминирующим развитие заболевания. Сами ранние эпилептические синдромы целесообразно рассматривать как неспецифическую реакцию незрелого мозга на экзо- или эндогенный стресс-фактор (возможно, один и тот же), реализующуюся эпилептическими припадками, тип которых зависит не от специфики фактора, а от степени структурно-функциональной состоятельности мозга.
Ранняя миоклоническая энцефалопатия. Ранняя миоклоническая энцефалопатия (РМЭ) — редкий возрастзависимый эпилептический синдром, впервые описанный J. Aicardi в 1978 г. В большинстве случаев заболевание начинается в возрасте, не превышающем 3 мес. Основным типом припадков являются миоклонии, преимущественно в виде фрагментарного мио-клонуса. Кроме того, могут наблюдаться частые внезапные парциальные приступы, массивные миоклонии и тонические спазмы.
Среди перечисленных клинических проявлений РМЭ патогномоничным признаком следует считать частые фрагментарные миоклонии, которые являются не только самым частым типом приступов, но и считаются дебютным, ранним симптомом заболевания. Тем не менее, с течением заболевания фрагментарные миоклонии постепенно уступают свою ведущую клиническую роль частым парциальным припадкам. Характеризуя особенности миоклонии, можно отметить, что они проявляются не только в состоянии бодрствования, но и во время сна. По степени выраженности они могут варьировать от легкого подергивания дистальных фаланг пальцев рук до миоклонии кистей, предплечий, век и угла рта. Частота их возникновения — от нескольких раз в день до нескольких десятков в минуту.
Парциальные припадки отмечаются приблизительно в 70—80 % случаев РМЭ и, подобно фрагментарным миоклониям, наблюдаются и во время бодрствования, и во время сна. Парциальные приступы при РМЭ отличаются большой частотой и могут достигать 300—500 раз в сутки.
К возрастному периоду 3—5 мес. супрессивно-взрывной ЭЭГ-паттерн постепенно замещается атипичной, или модифицированной, гипсаритмией, хотя в некоторых случаях он может персистировать достаточно долго.
Нейрорадиологические изменения при РМЭ не выражены. Как правило, и КТ, и МРТ не выявляют каких-либо грубых структурных изменений головного мозга. В тех редких случаях, когда они отмечаются, это преимущественно кортикальная атрофия различной степени выраженности.
Относительно этиологических аспектов РМЭ можно отметить, что не выявлено каких бы то ни было специфических этиологических факторов в развитии этого заболевания. Считается [Bernardina D., 1983], что определенную роль могут играть врожденные нарушения метаболизма (дизметабо-лические энцефалопатии раннего возраста), из которых выделяют как особо частые некетотическую гиперглицинемию, пропионовую ацидурию и D-глициновую ацидемию. В основном же большинство случаев РМЭ расценивается как криптогенные, т.е. формы, при которых презумптивно существующая причина, лежащая в основе развития эпилепсии, подразумевается, но не поддается идентификации на современном технологическом Уровне диагностических методов.
Лечение РМЭ составляет тяжелую и пока нерешенную проблему. К сожалению, к настоящему времени не существует антиконвульсантов и гормональных средств, которые могли бы обеспечить сколько-нибудь приемлемую эффективность лечения. Наиболее характерный исход заболевания смерть больных в первые 5 лет жизни; оставшиеся в живых страдают тяжелыми психомоторными расстройствами.
Ранняя эпилептическая энцефалопатия (синдром Отахары). Эта форма нцефалопатии является самым ранним по дебюту возрастзависимым эпилептическим синдромом. Она была впервые описана в 1978 г. японским ученым Shunsuke Ohtahara, а с 1989 г. признана в качестве самостоятельного эпилептического синдрома, получившего имя своего первооткрывателя — синдром Отахары.
В клинической картине приступы дебютируют в первые или 3 мес. жизни, но особенно часто в 1-й месяц. Основным типом припадков являются серийные или изолированные тонические спазмы. Приступы персистируют не только в состоянии бодрствования, но и ночью, помимо тонических спазмов, почти в половине случаев могут отмечаться моторные парциальные приступы, иногда по гемитипу. Миоклонические припадки нехарактерны, хотя в отдельных случаях могут иметь место.
Продолжительность тонического спазма при синдроме Отахары достигает приблизительно 10 с; в одной серии может отмечаться от 10 до 40 спазмов. Общее суточное количество спазмов достаточно велико и может достигать 300—400.
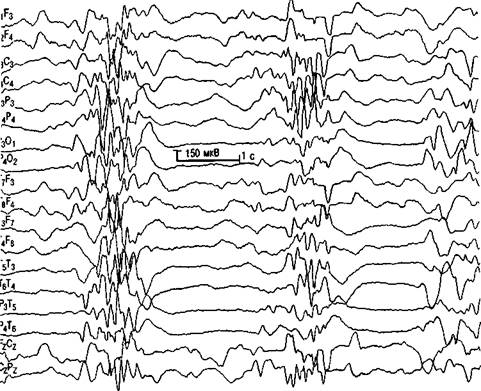
Основное ЭЭГ-проявление синдрома Отахары — описанный выше супрессивно-взрывной паттерн, который почти всегда отмечается и во сне, и в состоянии бодрствования. Вспышки медленных волн, длящиеся 1—3 с, имеют амплитуду 150—350 мкВ, перемежаются периодами почти полного уплощения ритма продолжительностью 3—4 с (рис. 3.9).

Рис. З.9. Ранняя инфантильная эпилептическая энцефалопатия (синдром Отахары). Диффузная супрессивно-взрывная активность.
В диагностике синдрома Отахары принципиально важны методы нейровизуализации, так как в отличие от РМЭ часто выявляют грубые структурные (порой асимметричные) изменения мозга. По данным S. Ohtahara (1997), эти нарушения отмечаются приблизительно в 85 % случаев ранней эпилептической энцефалопатии.
Как и РМЭ, синдром Отахары полиэтиологичен. В инициации заболевания особую роль играют врожденные мальформации головного мозга.
Мальформативные изменения мозга лежат в основе приблизительно 30—34 % случаев синдрома Веста [Jellinger К., 1987], по данным патоморфологических исследований. По своей классификационной сути это могут быть практически любые пороки развития мозга, включающие агенетические аномалии — агенезию мозолистого тела, агенетические порэнцефалические кисты, голопрозэнцефалию, агенезию червя и(или) гемисфер мозжечка; обширные эмбриоклас-тические процессы — гидранэнцефалию, эмбриокластические порэнцефалические кисты; гипо- и гиперпластические процессы — гипоплазию отдельных долей мозга, микроцефалию, унилатеральную мегалэнцефалию; кортикальные дисплазии — лиссэнцефалию, полимикрогирию, пахигирию, фокальную корковую дисплазию, ленточные и узловые нейронные гетеро-топии, микродисгенезии.
Особая роль в генезе инфантильных спазмов при синдроме Веста в последнее время уделяется микродисгенезиям, которые могут быть легко пропущены при рутинном диагностическом комплексе заболевания [Palm, 1986]. Предполагается, что небольшие участки дисплазированной ткани, Даже в небольшом количестве рассеянные в толще мозгового вещества, способны не только генерировать тяжелые и частые инфантильные спазмы, но и ответственны за грубые ментальные нарушения при синдроме Веста.
Ведущую роль в генезе инфантильных спазмов при этом заболевании безоговорочно отдают пери- и постнатальным гипоксически-ишемическим изменениям мозга [Chevrie, Aicardi, 1977].
Частота инфекционного фактора в развитии заболевания варьирует, по данным различных авторов, от 3 % [Matsumoto et , 1981] до И % [Lombroso C.T., 1983].
Инфекционные заболевания (цитомегалия, токсоплазмоз, герпетичес-й и краснушный энцефалиты, энтеровирусные и аденовирусные энцефалиты) могут играть более значимую роль в генезе инфантильных спазмов, так как вирусологические методы в настоящее время не позволяют проводить объективную оценку. Следует помнить, что свое катастрофическое эпилептогенное влияние они могут оказывать не напрямую, а опосредованно, например, через инициацию различных пороков развития.
Среди других (менее частых) причин возникновения синдрома Веста возмжно упомянуть нарушения метаболизма — до 10 % случаев [Meencke, srhard, 1985]; травматические изменения мозга (родовая травма); различие типы церебральных опухолей [Miyake et al., 1986].
Течение синдрома Веста в зависимости от этиологического фактора. В зависимости от этиологического фактора, лежащего в основе развития инфантильных спазмов при синдроме Веста, можно выделить несколько наиболее типичных типов течения заболевания:
• дальнейшее персистирование инфантильных спазмов в течение нескольких лет. Этот вариант течения заболевания чаще встречается при глубоких нарушениях кортикальной организации (особенно при диффузной лиссэнцефалии) или обширных эмбриокластических процессах (гидранэнцефалии);
• трансформация спазмов в мультифокальную эпилепсию. Как правило, такая процессуальность спазмов отмечается при множественных эпилептогенных нарушениях коры, например при туберозном склерозе или мультифокальной гипоксически-ишемической энцефалопатии. Результатом является появление мультифокальных или вторично-генерализованных приступов, относительно резистентных к антиком вульсантной терапии;
• трансформация спазмов в парциальную эпилепсию. Основные причины подобной трансформации включают порэнцефалию, очаговые формы кортикальных дисплазий и некоторые случаи туберозного склероза с единственным кортикальным туберсом;
• эволюция синдрома Веста в синдром Леннокса—Гасто. Это в основном отмечается при криптогенных инфантильных спазмах, т.е. спазмах, при которых этиологический фактор, лежащий в их основе, не идентифицируется, но подразумевается. Вообще отсутствие явных морфологических изменений мозга при столь губительной для прогноза трансформации инфантильных спазмов в синдром Леннокса— Гасто является одним из удивительных фактов эпилептологии, не поддающихся сколько-нибудь вразумительным объяснениям;
• полное спонтанное прекращение приступов. В подавляющем большинстве случаев отмечается у больных с криптогенными инфантильными спазмами.
Семиология эпилептических приступов при синдроме Веста.
Как уже говорилось, основным и единственным типом припадков при синдроме Веста являются инфантильные спазмы. Это один из кардинальных отличительных признаков заболевания от других ранних эпилептических энцефалопатий, в частности ранней миоклонической энцефалопатии. Спазмы представляют собой массивные генерализованные миоклонические или тонические сокращения аксиальной и конечностной мускулатуры.
Различают симметричные и асимметричные спазмы. При последних кинематика спазма имеет принципиально другой характер; наряду с типичными компонентами миоклонического или тонического инфантильного спазма отмечаются явные проявления парциального приступа, наиболее часто мимикрирующего асимметричный шейно-тонический рефлекс.
Продолжительность изолированного инфантильного спазма достаточно вариабельна и могут длиться от 0,5 до 2 сек.
Электроэнцефалографические аспекты синдрома Веста.
Основным (но не единственным) ЭЭГ-паттерном синдрома Веста является гипсаритмия. Гипсаритмия — это хаос и анархия на ЭЭГ.
Типичная гипсаритмия - это выраженное диффузное замедление активности, наличие диффузной быстрой активности; асимметрия фоновой записи.
При трансформации синдром Веста в синдром Леннокса—Гасто типичная функциональная гипсаритмия постепенно и неуклонно замещается модифицированной с преобладаем межполушарной синхронизации или генерализованной медленноволновой активности.

Рис. 3.10. ЭЭГ ребенка 5 мес с инфантильными спазмами. Модифицированная гипсаритмия.

Ранняя миоклоническая энцефалопатия – тяжелая форма эпилептической энцефалопатии, дебютирующая в возрасте до 3 месяцев, с высокой вероятностью летального исхода. Проявляется эпилептическим миоклонусом – практически постоянными мигрирующими подергиваниями отдельных мышц, чаще лица и конечностей. Характерна значительная задержка умственного развития. Диагностируется при помощи ЭЭГ-исследования, на энцефалограмме наблюдаются типичные изменения. Ранняя миоклоническая энцефалопатия резистентна к терапии противоэпилептическими препаратами, прогноз неблагоприятный.

- Причины ранней миоклонической энцефалопатии
- Симптомы ранней миоклонической энцефалопатии
- Диагностика и лечение ранней миоклонической энцефалопатии
- Цены на лечение
Общие сведения
Неонатальная миоклоническая энцефалопатия – одно из первых названий заболевания, предложенное неврологом Айкарди в 1978 году. С 1989 года патология классифицируется как форма симптоматической эпилепсии с неясной этиологией, относится к возрастзависимым синдромам. Частота встречаемости ранней миоклонической энцефалопатии составляет около 0,17%. В специализированной литературе описано всего около 100 случаев заболевания, однако многие дети не доживают до момента инструментальной диагностики, так что истинная распространенность, вероятно, выше. Патология с одинаковой частотой диагностируется у мальчиков и девочек. Несмотря на довольно редкие случаи выявления заболевания, в педиатрии оно считается крайне опасным, обеспечивающим высокий уровень летальности. Врачи при этом остаются бессильными и могут только попытаться поддержать жизнеспособность ребенка.
Причины ранней миоклонической энцефалопатии
Этиология заболевания по сей день остается не до конца изученной. Доказано влияние метаболических синдромов на развитие ранней миоклонической энцефалопатии. У заболевших детей часто диагностируются различные ацидемии, некетотическая гиперглицинемия, дефицит кофактора молибдена и другие врожденные дефекты метаболизма. Также выявлена некоторая связь с гипоксически-ишемическим поражением головного мозга, внутриутробными инфекциями и патологиями родов, однако данные факторы не всегда имеют место. В некоторых случаях установлено аутосомно-рецессивное наследование.
Ранняя миоклоническая энцефалопатия характеризуется отсутствием каких-либо структурных изменений головного мозга. Основная роль в патогенезе принадлежит нарушению процессов апоптоза глиальных клеток, в основном – астроцитов. Следствием является их скопление вдоль нейрональных аксонов. Предполагается, что именно этим обусловлена типичная клиническая картина заболевания. У большинства детей с рождения выявляется некоторая степень атрофии мозга, характерна задержка процессов миелинизации, что также может влиять на развитие симптомов ранней миоклонической энцефалопатии.
Симптомы ранней миоклонической энцефалопатии
Ранняя миоклоническая энцефалопатия может проявляться так называемыми инфантильными спазмами. Мозг новорожденного устроен таким образом, что развитие классических тонических приступов невозможно, и в данном случае наблюдаются диффузные тонические сокращения. В дальнейшем возможна трансформация в тонические приступы. В целом для ранней миоклонической энцефалопатии характерна высокая степень задержки умственного развития вплоть до вегетативного состояния (идиотия). Часто с рождения имеют место различные неврологические нарушения, в т. ч. нарушения мышечного тонуса новорожденных (гипертонус конечностей или мышечная гипотония), но иногда нервно-психическое состояние начинает прогрессивно ухудшаться только с появлением симптоматики энцефалопатии.
Диагностика и лечение ранней миоклонической энцефалопатии
Federico Vigevano
История и терминология
Ранняя миоклоническая энцефалопатия начинается с хаотических или фрагментарных спазмов. Могут наблюдаться и другие типы приступов – простые парциальные, массивная миоклония или тонические спазмы.
Вначале возникает хаотический парциальный миоклонус, который может наблюдаться в первые часы после рождения. Миоклонус обычно захватывает конечности и лицо, может ограничиться подергиваниями брови, губ или пальцев. Судороги появляются во время сна или после пробуждения, их часто называют хаотическими за то, что они быстро и асинхронно переходят от одной части тела к другой, с частотой от единичных до почти непрерывных. В некоторых случаях к ограниченному парциальному миоклонусу могут добавиться и генерализованные миоклонии. После хаотического миоклонуса часто появляются короткие парциальные приступы. Проявления парциальных приступов едва уловимые, например, только девиация глазных яблок или вегетативные феномены (апноэ или покраснение лица) (Dalla Bernardina et al 1983). Тонические приступы, которые описываются довольно часто, появляются на первом месяце жизни или позже, возникают в состоянии сна или бодрствования (Aicardi and Ohtahara 2002). Клинически у ребенка появляются диффузные тонические сокращения, распространяющиеся на конечности. Настоящие эпилептические спазмы редки и появляются позже. Неврологический статус типичен: выраженное задержка психомоторного развития, заметная гипотония, повышенная активация, иногда вегетативное состояние (Aicardi 1992). Описываемые признаки дальнейшего ухудшения состояния (Dalla Bernardina et al 1983) трудно документировать из-за раннего дебюта заболевания. Изредка развивается симптоматика периферической полинейропатии.
В описанных в литературе случаях ни разу не отмечались акушерские или какие-либо другие перинатальные осложнения. Соответственно, считается, что ранняя миоклоническая энцефалопатия различна может иметь различную этиологию. Сиблинги заболевают очень редко (Dalla Bernardina et al 1983; Aicardi 1992; Wang et al 1998), среди родителей не отмечалось близких родственников, и они были предположительно здоровы. Наиболее вероятным является аутосомно-рецессивное наследование, но и оно не доказано.
Патогенез и патофизиология
Отсутствие постоянных общих патоморфологических изменений позволяет предположить полиэтиологичность заболевания. Среди патологических изменений часто встречаются кортиконейрональная гетеротопия и пролиферация астроцитов, порэнцефалия, периваскулярные концентрические тела, демиелинизация в полушариях мозга, лиссэнцефалия, односторонняя мегалоцефалия с пролиферацией астроцитов (Aicardi 1985). С другой стороны, описаны 2 пациента, у которых не было обнаружено никаких патоморфологических изменений (Dalla Bernardina et al 1983).
Несмотря на разные причины развития заболевания, Spreafico at al. предположили наличие общего патологического звена: огромное количество астроцитов вдоль аксонов в белом веществе извилин головного мозга расценивается как нарушение процессов апоптоза глиальных клеток в процессе развития и созревания головного мозга (Spreafico et al 1993).
Ранняя миоклоническая энцефалопатия очень редкое заболевание. Статистические исследования детской эпилепсии, проведенные в префектуре Окаяма в Японии, обнаружили 4 случая ранней миоклонической энцефалопатии (0,168%) среди 2378 пациентов с эпилепсией, моложе 10 лет на момент 31 декабря 1980г (Oka et al 1995). Распространенность ранней миоклонической энцефалопатии была выше, чем синдрома Отахара (0,04%), но значительно ниже, чем синдрома Веста (1.68%). Не так давно было получены похожие данные по распространенности этого заболевания в том же регионе (Oka 2002).
Развитие хаотического миоклонуса и отсутствие тонических спазмов позволяет отличать раннюю миоклоническую энцефалопатию от синдрома Отахара. При синдроме Отахара паттерн ЭЭГ отличается более продолжительными вспышками и более короткими периодами подавления. Этиологически синдром Отахара всегда связан со структурными нарушениями мозга, тогда как в ряде случаев ранней миоклонической энцефалопатии мы находим метаболические нарушения, высок доля криптогенных случаев. Прогноз при ранней миоклонической энцефалопатии менее благоприятен.
Хаотический миоклонус не всегда сопровождается на ЭЭГ эпилептиформными изменениями. Изменения на ЭЭГ при парциальных приступах похожи на те же, что и при неонатальных судорогах. Данные нейровизуализации различны и зависят от этиологии: от нормы до гемимегалоцефалии, дилатации боковых желудочков или кортикальной и перивентрикулярной атрофии (Aicardi 1985).
Учитывая вышеописанные врожденные нарушения метаболизма мозга, необходимо исследовать содержание аминокислот в сыворотке крови, таких как глицин и его метаболиты, органические кислоты и аминокислоты в цереброспинальной жидкости.
Прогноз и осложнения
При ранней миоклонической энцефалопатии прогноз очень неблагоприятный. Летальность во всех описанных случаях составляла более 50% (такие пациенты умирали на 1-2 году жизни), у многих пациентов развивалось вегетативное состояние. Ранняя миоклоническая энцефалопатия может сохраняться и в более старшем возрасте или эволюционировать в тяжелую парциальную эпилепсию.
На сегодняшний день не существует эффективного лечения ранней миоклонической энцефалопатии. Ни антиэпилептические препараты, ни стероиды, ни АКТГ не влияют практически на прогноз. При некетоновой гиперглицинемии пиридоксин и бензоат могут нормализовать уровень глицина в крови и улучшить ЭЭГ-картину, но это никак не скажется на прогнозе заболевания. Проба с пиридоксином диагностически оправдана во всех случаях ранней миоклонической энцефалопатии.
Список литературы
Aicardi J. Early myoclonic encephalopathy. In: Roger J, Dravet C, Bureau M, Dreifuss FE, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey Eurotext, 1985.
Aicardi J. Early myoclonic encephalopathy (neonatal myoclonic encephalopathy). In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 2nd ed. London: John Libbey, 1992:13-23.
Aicardi J, Goutieres F. Encephalopathie myoclonique neonatale. Rev EEG Neurophysiol 1978;8:99-101.
Aicardi J, Ohtahara S. Severe neonatal epilepsies with suppression-burst pattern. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London: John Libbey, 2002:33-44.
Aukett A, Bennett MJ, Hosking GP. Molybdenum cofactor deficiency: an easily missed inborn error of metabolism. Dev Med Child Neurol 1988;30: 531-5.
Cavazzuti GB, Nalin A, Ferrari F, Grandori L, Beghini GE. Encefalopatia epilettica ad insorgenza neonatale. Clin Pediatr 1978;60:239-46.
Commission of Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
Dalla Bernardina B, Aicardi J, Goutieres F, Plouin P. Glycine encephalopathy. Neuropadiatrie 1979;10:209-25.
Dalla Bernardina B, Dulac O, Fejerman N, et al. Early myoclonic epileptic encephalopathy (E.M.E.E.). Eur J Pediatr 1983;140:248-52.
Engel J Jr; International League Against Epilepsy. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001;42:796-803.
Grandgeorge D, Favier A, Bost M, et al. L'acidemie D-glycerique. A propos d'une nouvelle observation anatomo-clinique. Arch Franc Pediatr 1980;37:577-84.
Lombroso C. Early myoclonic encephalopathy, early infantile epileptic encephalopathy and benign and severe infantile myoclonic epilepsies: a critical review and personal contributions. J Clin Neurophysiol 1990;7:380-408.
Martin HJ, Deroubaix-Tela P, Thelliez P. Encephalopathie epileptique neonatale a bouffees periodiques. Rev EEG Neurophysiol Clin 1981;11:397-403.
Ohtahara S, Ohtsuka Y, Erba G. Early epileptic encephalopathy with suppression-burst. In: Engel J Jr, Pedley T, editors. Epilepsy: A comprehensive textbook. Vol. 3. Philadelphia: Lippincott-Raven, 1998:2257-61.
Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neurophysiol 2003;20:398-407.
Oka E. Childhood epilepsy in Okayama Prefecture, Japan: a neuroepidemiological study. No To Hattatsu (Tokyo) 2002;34:95-102.
Oka E, Ishida S, Ohtsuka Y, Ohtahara S. Neuroepidemiological study of childhood epilepsy by application of international classification of epilepsies and epileptic syndromes (ILAE, 1989). Epilepsia 1995;36:658-61.
Schlumberger E, Dulac O, Plouin P. Early infantile syndrome(s) with suppression-burst: nosological considerations: In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes of infancy, childhood and adolescence. 2nd ed. London: John Libbey, 1992:35-42.
Spreafico R, Angelini L, Binelli S, et al. Burst suppression and impairment of neocortical ontogenesis: electroclinical and neuropathologic findings in two infants with early myoclonic encephalopathy. Epilepsia 1993;34(5):800-8.
Vigevano F, Bartuli A. Infantile epileptic syndromes and metabolic etiologies. J Child Neurology 2002;17(3):3S9-13.
Vigevano F, Bosman C, Gisondi A, Maccagnani F, Seganti G, Sergo M. Neonatal myoclonic epileptic encephalopathy without hyperglycinemia. Electroencephal Clin Neurophysiol 1981;52:52P-3P.
Vigevano F, Maccagnani F, Bertini E, et al. Encefalopatia mioclonica precoce associata ad alti livelli di acido propionico nel siero. Boll Lega Ital Epil 1982;39:181-2.
Wang PJ, Lee WT, Hwu C, et al. The controversy regarding diagnostic criteria for early myoclonic encephalopathy. Brain Dev 1998;20:530-5.
Читайте также:
