
Синдром псевдо-леннокса атипичная фокальная эпилепсия детского возраста
Этиология. Предположительно идиопатическая форма эпилепсии. В 5% случаев роландическая эпилепсия может трансформироваться в синдром псевдо-Леннокса (СПЛ). Данная форма эпилепсии признана большинством детских эпилептологов, но пока еще не введена ни в одну международную классификацию.
Диагностические критерии. Заболевание впервые было описано Aicardi & Chevrie в 1982 году. СПЛ дебютирует в дошкольном возрасте, преимущественно, от 1,5 до 6 лет. Для СПЛ характерен полиморфизм приступов:
- фокальные моторные,
- атонические (негативный миоклонус),
- атипичные абсансы,
- вторично - генерализованные тонико – клонические,
- миоклонические (активный миоклонус),
- эпилептический статус.
Основной вид приступов - короткие фокальные моторные в виде фаринго – оральных, гемифациальных или фацио - брахиальных пароксизмов с нарушением речи, гиперсаливацией, возникающие при засыпании или пробуждении. Этот тип приступов характерен для роландической эпилепсии и наблюдается обычно в дебюте заболевания. Далее частота приступов катастрофически нарастает; присоединяются атонические пароксизмы и атипичные абсансы. Фокальные атонические приступы (негативный миоклонус) проявляются в виде пассивных кивков, наклонов туловища, ступенчатых приседаний, и наконец, мгновенных падений без судорог. Частота данных приступов достигает нескольких десятков в сутки, нарастая в период после пробуждения пациентов. Вторично - генерализованные судорожные приступы (обычно ассоциированные со сном) и миоклонические пароксизмы (активный миоклонус) встречаются у 1/3 больных СПЛ.
При неадекватном лечении нередко возникает эпилептический статус атипичных абсансов или фаринго – оральных приступов со снижением двигательной (ступор) и психической активности, анартрией, гиперсаливацией, атоническими феноменами (кивки, наклоны). Статус чаще наступает в утреннее время (после пробуждения) и может продолжаться часами.
Неврологическое обследование нередко выявляет динамическую атаксию, интенционный тремор, дисметрию, брадикинезию, скандированную речь. Выраженность данных симптомов флюктуирует, нарастая в период учащения приступов. Во время активного периода заболевания у всех пациентов констатируются выраженные когнитивные и речевые расстройства, резко затрудняющие их обучение [Hahn, 2000].
ЭЭГ при СПЛ характеризуется сочетанием региональной и диффузной эпилептиформной активности. Основной ЭЭГ паттерн – высокоамплитудная эпилептиформная активность острая – медленная волна (идентичная по морфологии доброкачественным эпилептиформным паттернам детства), возникающая, преимущественно, в передне - центральных отведениях и имеющая высокий индекс представленности. Характерно усиление эпилептиформной активности в медленном сне с появлением продолженной диффузной активности, возникающей в результате феномена вторичной билатеральной синхронизации. Результаты, полученные при нейровизуализации, неспецифичны. Возможно появление умеренной кортикальной и субкортикальной атрофии. Локальные нарушения, как правило, отсутствуют.
Определение инвалидности при СПЛ следует проводить с осторожностью и решать индивидуально. Во время активного периода заболевания пациенты обычно не способны посещать массовую школу ввиду высокой частоты приступов и выраженных когнитивных нарушений. После 10 лет активность заболевания уменьшается, однако, у ряда больных когнитивные и речевые расстройства остаются, что существенно снижает их способность к обучению и социальную адаптацию.
Терапия. Стартовое лечение начинается с производных вальпроевой кислоты. Назначается конвульсофин в дозе 900-2000 мг/сут (30-70 мг/кг/сут) в 2-3 приема. Вальпроаты особенно эффективны при миоклонических, генерализованных судорожных приступах и атипичных абсансах.
Препарат второго выбора – топирамат. Топамакс назначается с постепенным увеличением дозы до 75-200 мг/сут (3-7 мг/кг/сут) в 2 приема. Он эффективен при фокальных моторных, генерализованных судорожных и атонических приступах. В единичных случаях высокие дозы препарата могут приводить к парадоксальному учащению приступов.
Препарат третьего выбора – леветирацетам. Кеппра назначается с постепенной титрацией дозы до 750-2500 мг/сут (30-60 мг/кг/сут) в 2 приема. Препарат эффективен при фокальных моторных, генерализованных судорожных и миоклонических приступах. Hoppen и соавт. (2003) показали высокую эффективность кеппры в средней дозе 50 мг/кг/сут у больных синдромом псевдо-Леннокса в комбинации с вальпроатами.
При неэффективности монотерапии следует переходить к комбинированному лечению. Оптимальные комбинации в лечении СПЛ: вальпроаты + сукцинимиды, вальпроаты + топирамат, вальпроаты + леветирацетам, а также указанные препараты в комбинации с бензодиазепинами. Присоединять суксилеп к базовым АЭП рекомендуется сразу после неэффективности монотерапии. Суксилеп назначается в дозе 500-1000 мг/сут (20-35 мг/кг/сут) в 3 приема. Такая комбинация особенно эффективна в отношении псевдогенерализованных приступов (атонические, миоклонические, атипичные абсансы) и продолженной диффузной эпилептиформной активности на ЭЭГ.
Следующая комбинация – вальпроаты, топирамат или леветирацетам + бензодиазепины. Назначается фризиум в дозе 10-30 мг/сут (0,5-1,0 мг/кг/сут) в 2-3 приема. Нередко добавление фризиума приводит к резкому урежению приступов и улучшению когнитивных функций. Однако обычно этот эффект кратковременный. Фризиум рекомендуется в период учащения приступов, а также при развитии эпилептического статуса фокальных моторных приступов или атипичных абсансов.
Применение препаратов карбамазепина и барбитуратов не рекомендуется ввиду возможности учащения приступов.
При отсутствии эффективности от АЭП назначаются стероидные гормоны (синактен - депо, преднизолон, метипред, дексаметазон). Синактен – депо назначается, начиная с 0,1 мг внутримышечно 1 раз в сутки с постепенным наращиванием по 0,1 мг 1 раз в 2-5 дней до дозы 1,0 мг в сутки. Продолжительность терапии - 1-2 мес. с постепенным снижением. Средняя доза преднизолона – 2 мг/кг/сут перорально однократно утром. Возможно применение дексаметазона по схеме: дексаметазон в дозе 2 мг/кг/сут перорально в течение 1 нед., затем 1 мг/кг/сут еще 2 нед., затем переход на альтернирующий метод терапии – 1 поддерживающая доза (например, 0,5 мг/кг/сут) раз в 2-3 дня в течение 3-6 мес.
Прогноз при СПЛ следует проводить с осторожностью. Во время активного периода заболевания приступы обычно резистентны к проводимой терапии. В связи с этим, необходимы высокие дозы препаратов, нередко с включением бензодиазепинов и гормонов. После 9-и летнего возраста частота приступов постепенно снижается; после 11 лет постепенно блокируется эпилептиформная активность; и к началу пубертатного периода пациенты достигают полной электро – клинической ремиссии [Мухин К.Ю. и соавт., 2001; Fejerman и соавт., 2000]. Выраженность когнитивных нарушений также уменьшается с возрастом. Однако у ряда больных когнитивные и речевые расстройства остаются, не смотря на прекращение приступов, что существенно снижает их способность к обучению и социальную адаптацию.
Полный текст:
- Аннотация
- Об авторе
- Список литературы
- Cited By
1. Миронов М.Б., Мухин К.Ю., Какаулина В.С. Аггравация эпилептического негативного миоклонуса у детей при назначении карбамазепина и окскарбазепина. Фарматека 2012;(1):67–71. [Мironov М.B., Мukhin K.Yu., Kakaulina V.S. Аggravation of the epileptic negative myoclonus at children, receiving carbamazepine and oxcarbazepine. Farmateka = Pharmateca 2012;(1):67–71. (In Russ.)].
2. Мухин К.Ю. Синдром псевдоленнокса. В кн.: Эпилепсия: атлас электро-клинической диагностики. Под ред. К.Ю. Мухина, А.С. Петрухина, Л.Ю. Глуховой. М.: Альварес Паблишинг, 2004. С. 322–36. [Мukhin K.Yu. Pseudo-Lennox syndrome. In: Epilepsy: аtlas of the electric & clinical diagnostics. Ed. by K.Yu. Мukhin, А.S. Petrukhin, L.Yu. Glukhova. Мoscow: Аl’vares Pablishing, 2004. Pp. 322–36. (In Russ.)].
3. Мухин К.Ю. Синдром псевдо-Леннокса (атипичная доброкачественная парциальная эпилепсия детского возраста). В кн.: Мухин К.Ю., Петрухин А.С., Холин А.А. Эпилептические энцефалопатии и схожие синдромы у детей. М.: АртСервис ЛТД, 2011. С. 322–42. [Мuhin K.Yu. PseudoLennox sydnrome (аtypical benign partial childhood epilepsy). In: Мukhin K.Yu., Petrukhin А.S., Kholin А.А. Epileptic encephalopathies and related syndromes at children. Мoscow: АrtServis LTD, 2011. Pp. 322–42. (In Russ.)].
4. Мухин К.Ю., Глухова Л.Ю., Петрухин А.С. и др. Диагностические критерии синдрома атипичной доброкачественной парциальной эпилепсии детского возраста. Журнал неврологии и психиатрии им. С.С. Корсакова 2001;101(1):13–21. [Мukhin K.Yu., Glukhovа L.Yu., Petrukhin А.S. et al. Diagnostic criteria of the atypical benign childhood epilepsy. Zhurnal nevrologii i psikhiatrii im. S.S. Korsakova = S.S. Korsakov Journal of Neurology and Psychiatry 2001;101(1):13–21. (In Russ.)].
5. Ноговицын В.Ю., Нестеровский Ю.Е., Осипова Г.Н. и др. Полиморфизм электроэнцефалографического паттерна доброкачественных эпилептиформных нарушений в детстве. Журнал неврологии и психиатрии им. С.С. Корсакова 2004;104(10): 48–56. [Nogovitsyn V.Yu., Nesterovskiy Yu.Е., Оsipovа G.N. et al. Polymorphism of the electric encephalographic pattern of benign epileptiform discharges of childhood. Zhurnal nevrologii i psikhiatrii im. S.S. Korsakova = S.S. Korsakov Journal of Neurology and Psychiatry 2004;104(10):48–56. (In Russ.)].
6. Aicardi J., Chevrie J.J. Atypical benign partial epilepsy of childhood. Dev Med Child Neurol 1982;24(3):281–92.
7. Beaumanoir A., Mira L. Secondary bilateral synchrony: significant EEG pattern in frontal lobe seizures. In: Frontal lobe seizures and epilepsies in children. Ed. by A. Beaumanoir, F. Andermann, P. Chauvel et al. Paris: John Libbey Eurotext, 2003. Pp. 195–205.
8. Boenigk H.E. Personal communication. Bielefeld, 2000.
9. Capovilla G., Beccaria F., Bianchi A. et al. Ictal EEG patterns in epilepsy with centrotemporal spikes. Brain Dev 2011;33(4):301–9.
10. Dalla-Bernardina B., Sgro V., Fejerman N. Epilepsy with centro-temporal spikes and related syndromes. In: Epileptic syndromes in infancy, childhood and adolescence. Ed. by J. Roger, M. Bureau, Ch. Dravet et al. 4th edn. John Libbey Eurotext, 2005. Pp. 203–25.
11. Dalla-Bernardina B., Tassinari C.A. EEG of a nocturnal seizure in a patient with “benign epilepsy of childhood with rolandic spikes”. Epilepsia 1975;16(3):497–501.
12. De Bellescize J., Specchio N., Arzimanoglou A. “Benign” epilepsies in infants: are they always benign? In: Seizures and syndromes of onset in the two first years of life. Ed. by S. Moshe, H. Cross, J. de Bellescize et al. John Libbey Eurotext, 2015. Pp. 185–204.
13. Deonna T., Ziegler H.L., Despland P.A. Combined myoclonic-astatic and “benign” focal epilepsy of childhood (“atypical benign partial epilepsy of childhood”). A separate syndrome? Neuropediatrics 1986;17(3): 144–51.
14. Dittrich C., Diener W., Hahn A. et al. Benign course in 68 patients with epilepsy initially simulating Lennox syndrome – pseudo-Lennox syndrom. Epilepsia 1999;40(Suppl 2):168.
15. Doose H.P. EEG in childhood epilepsy. Hamburg: John Libbey, 2003. Pp. 202–10.
16. Doose H.P., Baier W.K. Benign partial epilepsy and related conditions: multifactorial pathogenesis with hereditary impairment of brain maturation. Eur J Pediatr 1989;149(3):152–8.
17. Doose H., Neubauer B.A., Petersen B. The concept of hereditary impairment of brain maturation. Epileptic Disord 2000;2(Suppl 1):45–9.
18. Engel J. Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001;42(6):796–803.
19. Fejerman N. Atypical rolandic epilepsy. Epilepsia 2009;50(Suppl 7):9–12.
20. Fejerman N., Caraballo R., DallaBernardina B. Atypical evolution of benign focal epilepsies in childhood. In: Benign focal epilepsies in infancy, childhood and adolescence. Ed. by N. Fejerman, R. Caraballo. John Libbey Eurotext, 2007. Pp. 179–219.
21. Fejerman N., Caraballo R., Tenembaum S. Atypical evolutions of benign localization – related epilepsies in children: are they predictable? Epilepsia 2000;41(4):380–90.
22. Fejerman N., Di Blasi A.M. Status epilepticus of benign partial epilepsies in children: report of two cases. Epilepsia 1987;28(4):351–5.
23. Gobbi G., Boni A., Filippini M. The spectrum of idiopathic rolandic epilepsy syndromes and occipital epilepsies: from the benign to the disabling. Epilepsia 2006;47(Suppl 2):62–6.
24. Gobbi G., Grosso S. Atypical benign partial epilepsy of childhood. In: Atlas of epilepsies. Ed. by C. Panayiotopoulos. London: Springer, 2010. Pp. 923–30.
25. Gross-Selbeck G. Treatment of “benign” partial epilepsies of childhood, including atypical forms. Neuropediatrics 1995;26(1):45–50.
26. Guerrini R., Belmonte A., Genton P. Antiepileptic drugs-induced worsening of seizures in children. Epilepsia 1998; 39(Suppl 3):2–10.
27. Hahn A. Atypical benign partial epilepsy/ pseudo-Lennox syndrome. Epileptic Disord 2000;2(Suppl 1):11–7.
28. Hahn A., Pistohl J., Neubauer B. Clinical features and outcome of atypical benign partial epilepsy (pseudo-Lennox syndrome). Epilepsie Blatter 1997;10:56.
29. Hahn A., Pistohl J., Neubauer B., Stephani U. Atypical “benign” partial epilepsy or pseudo-Lennox syndrome. Part I: Symptomatology and long-term prognosis. Neuropediatrics 2001;32(1):1–8.
30. Kelemen A., Barsi P., Gyorzok Z. et al. Thalamic lesion and epilepsy with generalized seizures, ESES and spike-wave paroxysms – report of three cases. Seizure 2006;15(6): 454–8.
31. Kramer U. Atypical presentations of benign childhood epilepsy with centrotemporal spikes: a review. J Child Neurol 2008;23(7):785–90.
32. Matsuoka H., Nakamura M., Ohno T. et al. The role of cognitive-motor function in precipitation and inhibition of epileptic seizures. Epilepsia 2005;46(Suppl 1): 17–20.
33. Michelucci R., Rizzi R., Passarelli D. et al. Ictal “sleep” as a sole manifestation of partial status epilepticus: video-EEG recording of one patient. Boll Lega It Epil 1998;102/103:223–4.
34. Mukhin K.Yu., Glukhova L.Yu., Petrukhin A.S. et al. Diagnostic criteria in atypical benign partial epilepsy of childhood. Search Epil 2002;3:20–1.
35. Oguni H., Fukuyama Y., Imaizumi Y., Uehara T. Video-EEG analysis of drop seizures in myoclonic-astatic epilepsy of early childhood (Doose syndrome). Epilepsia 1992;33(5):805–13.
36. Panayiotopoulos C., Bureau M., Caraballo R. et al. Idiopathic focal epilepsies in children. In: Epileptic syndromes in infancy, childhood and adolescence. Ed. by M. Bureau, P. Genton, Ch. Dravet et al. 5th edn. with video. Paris: John Libbey Eurotext, 2012. Pp. 217–54.
37. Roulet E., Deonna T., Despland P.A. Prolonged intermittent drooling and oromotor dyspraxia in benign childhood epilepsy with centrotemporal spikes. Epilepsia 1989;30(5):564–8.
38. Rubboli G., Tassinari C.A. Negative myoclonus. An overview of its clinical features, pathophysiological mechanisms and management. Neurophysiol Clin 2006; 36(5–6):337–43.
39. Stephani U. Typical semiology of benign childhood epilepsy with centrotemporal spikes (BCECTS). Epileptic Disord 2000; 2(Suppl 1):3–4.
40. Tassinari C.A., Rubboli G., Shibasaki H. Neurophysiology of positive and negative myoclonus. Electroencephalogr Clin Neurophysiol 1998;107(3):181–95.

Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Синдром псевдо-Леннокса (СПЛ) впервые был описан Aicardi & Chevrie в 1982 году. СПЛ дебютирует в дошкольном возрасте, преимущественно, от 1,5 до 6 лет. Для СПЛ характерен полиморфизм приступов:
- фокальные моторные,
- атонические (негативный миоклонус),
- атипичные абсансы,
- вторично — генерализованные тонико – клонические,
- миоклонические (активный миоклонус),
- эпилептический статус.
Основной вид приступов — короткие фокальные моторные в виде фаринго – оральных, гемифациальных или фацио — брахиальных пароксизмов с нарушением речи, гиперсаливацией, возникающие при засыпании или пробуждении. Этот тип приступов характерен для роландической эпилепсии и наблюдается обычно в дебюте заболевания. Далее частота приступов катастрофически нарастает; присоединяются атонические пароксизмы и атипичные абсансы. Фокальные атонические приступы (негативный миоклонус) проявляются в виде пассивных кивков, наклонов туловища, ступенчатых приседаний, и наконец, мгновенных падений без судорог. Частота данных приступов достигает нескольких десятков в сутки, нарастая в период после пробуждения пациентов. Вторично — генерализованные судорожные приступы (обычно ассоциированные со сном) и миоклонические пароксизмы (активный миоклонус) встречаются у 1/3 больных СПЛ.
При неадекватном лечении нередко возникает эпилептический статус атипичных абсансов или фаринго – оральных приступов со снижением двигательной (ступор) и психической активности, анартрией, гиперсаливацией, атоническими феноменами (кивки, наклоны). Статус чаще наступает в утреннее время (после пробуждения) и может продолжаться часами.
Неврологическое обследование нередко выявляет динамическую атаксию, интенционный тремор, дисметрию, брадикинезию, скандированную речь. Выраженность данных симптомов флюктуирует, нарастая в период учащения приступов. Во время активного периода заболевания у всех пациентов констатируются выраженные когнитивные и речевые расстройства, резко затрудняющие их обучение [Hahn, 2000].
ЭЭГ при СПЛ характеризуется сочетанием региональной и диффузной эпилептиформной активности. Основной ЭЭГ паттерн – высокоамплитудная эпилептиформная активность острая – медленная волна (идентичная по морфологии доброкачественным эпилептиформным паттернам детства), возникающая, преимущественно, в передне — центральных отведениях и имеющая высокий индекс представленности. Характерно усиление эпилептиформной активности в медленном сне с появлением продолженной диффузной активности, возникающей в результате феномена вторичной билатеральной синхронизации. Результаты, полученные при нейровизуализации, неспецифичны. Возможно появление умеренной кортикальной и субкортикальной атрофии. Локальные нарушения, как правило, отсутствуют.
Определение инвалидности при СПЛ следует проводить с осторожностью и решать индивидуально. Во время активного периода заболевания пациенты обычно не способны посещать массовую школу ввиду высокой частоты приступов и выраженных когнитивных нарушений. После 10 лет активность заболевания уменьшается, однако, у ряда больных когнитивные и речевые расстройства остаются, что существенно снижает их способность к обучению и социальную адаптацию.
Этиология. Предположительно идиопатическая форма эпилепсии. В 5% случаев роландическая эпилепсия может трансформироваться в синдром псевдо-Леннокса (СПЛ). Данная форма эпилепсии признана большинством детских эпилептологов, но пока еще не введена ни в одну международную классификацию.
Диагностические критерии. Заболевание впервые было описано Aicardi & Chevrie в 1982 году. СПЛ дебютирует в дошкольном возрасте, преимущественно, от 1,5 до 6 лет. Для СПЛ характерен полиморфизм приступов:
- фокальные моторные,
- атонические (негативный миоклонус),
- атипичные абсансы,
- вторично — генерализованные тонико – клонические,
- миоклонические (активный миоклонус),
- эпилептический статус.
Основной вид приступов — короткие фокальные моторные в виде фаринго – оральных, гемифациальных или фацио — брахиальных пароксизмов с нарушением речи, гиперсаливацией, возникающие при засыпании или пробуждении. Этот тип приступов характерен для роландической эпилепсии и наблюдается обычно в дебюте заболевания. Далее частота приступов катастрофически нарастает; присоединяются атонические пароксизмы и атипичные абсансы. Фокальные атонические приступы (негативный миоклонус) проявляются в виде пассивных кивков, наклонов туловища, ступенчатых приседаний, и наконец, мгновенных падений без судорог. Частота данных приступов достигает нескольких десятков в сутки, нарастая в период после пробуждения пациентов. Вторично — генерализованные судорожные приступы (обычно ассоциированные со сном) и миоклонические пароксизмы (активный миоклонус) встречаются у 1/3 больных СПЛ.
При неадекватном лечении нередко возникает эпилептический статус атипичных абсансов или фаринго – оральных приступов со снижением двигательной (ступор) и психической активности, анартрией, гиперсаливацией, атоническими феноменами (кивки, наклоны). Статус чаще наступает в утреннее время (после пробуждения) и может продолжаться часами.
Неврологическое обследование нередко выявляет динамическую атаксию, интенционный тремор, дисметрию, брадикинезию, скандированную речь. Выраженность данных симптомов флюктуирует, нарастая в период учащения приступов. Во время активного периода заболевания у всех пациентов констатируются выраженные когнитивные и речевые расстройства, резко затрудняющие их обучение [Hahn, 2000].
ЭЭГ при СПЛ характеризуется сочетанием региональной и диффузной эпилептиформной активности. Основной ЭЭГ паттерн – высокоамплитудная эпилептиформная активность острая – медленная волна (идентичная по морфологии доброкачественным эпилептиформным паттернам детства), возникающая, преимущественно, в передне — центральных отведениях и имеющая высокий индекс представленности. Характерно усиление эпилептиформной активности в медленном сне с появлением продолженной диффузной активности, возникающей в результате феномена вторичной билатеральной синхронизации. Результаты, полученные при нейровизуализации, неспецифичны. Возможно появление умеренной кортикальной и субкортикальной атрофии. Локальные нарушения, как правило, отсутствуют.
Определение инвалидности при СПЛ следует проводить с осторожностью и решать индивидуально. Во время активного периода заболевания пациенты обычно не способны посещать массовую школу ввиду высокой частоты приступов и выраженных когнитивных нарушений. После 10 лет активность заболевания уменьшается, однако, у ряда больных когнитивные и речевые расстройства остаются, что существенно снижает их способность к обучению и социальную адаптацию.
Терапия. Стартовое лечение начинается с производных вальпроевой кислоты. Назначается конвульсофин в дозе 900-2000 мг/сут (30-70 мг/кг/сут) в 2-3 приема. Вальпроаты особенно эффективны при миоклонических, генерализованных судорожных приступах и атипичных абсансах.
Препарат второго выбора – топирамат. Топамакс назначается с постепенным увеличением дозы до 75-200 мг/сут (3-7 мг/кг/сут) в 2 приема. Он эффективен при фокальных моторных, генерализованных судорожных и атонических приступах. В единичных случаях высокие дозы препарата могут приводить к парадоксальному учащению приступов.
Препарат третьего выбора – леветирацетам. Кеппра назначается с постепенной титрацией дозы до 750-2500 мг/сут (30-60 мг/кг/сут) в 2 приема. Препарат эффективен при фокальных моторных, генерализованных судорожных и миоклонических приступах. Hoppen и соавт. (2003) показали высокую эффективность кеппры в средней дозе 50 мг/кг/сут у больных синдромом псевдо-Леннокса в комбинации с вальпроатами.
При неэффективности монотерапии следует переходить к комбинированному лечению. Оптимальные комбинации в лечении СПЛ: вальпроаты + сукцинимиды, вальпроаты + топирамат, вальпроаты + леветирацетам, а также указанные препараты в комбинации с бензодиазепинами. Присоединять суксилеп к базовым АЭП рекомендуется сразу после неэффективности монотерапии. Суксилеп назначается в дозе 500-1000 мг/сут (20-35 мг/кг/сут) в 3 приема. Такая комбинация особенно эффективна в отношении псевдогенерализованных приступов (атонические, миоклонические, атипичные абсансы) и продолженной диффузной эпилептиформной активности на ЭЭГ.
Следующая комбинация – вальпроаты, топирамат или леветирацетам + бензодиазепины. Назначается фризиум в дозе 10-30 мг/сут (0,5-1,0 мг/кг/сут) в 2-3 приема. Нередко добавление фризиума приводит к резкому урежению приступов и улучшению когнитивных функций. Однако обычно этот эффект кратковременный. Фризиум рекомендуется в период учащения приступов, а также при развитии эпилептического статуса фокальных моторных приступов или атипичных абсансов.
Применение препаратов карбамазепина и барбитуратов не рекомендуется ввиду возможности учащения приступов.
При отсутствии эффективности от АЭП назначаются стероидные гормоны (синактен — депо, преднизолон, метипред, дексаметазон). Синактен – депо назначается, начиная с 0,1 мг внутримышечно 1 раз в сутки с постепенным наращиванием по 0,1 мг 1 раз в 2-5 дней до дозы 1,0 мг в сутки. Продолжительность терапии — 1-2 мес. с постепенным снижением. Средняя доза преднизолона – 2 мг/кг/сут перорально однократно утром. Возможно применение дексаметазона по схеме: дексаметазон в дозе 2 мг/кг/сут перорально в течение 1 нед., затем 1 мг/кг/сут еще 2 нед., затем переход на альтернирующий метод терапии – 1 поддерживающая доза (например, 0,5 мг/кг/сут) раз в 2-3 дня в течение 3-6 мес.
Прогноз при СПЛ следует проводить с осторожностью. Во время активного периода заболевания приступы обычно резистентны к проводимой терапии. В связи с этим, необходимы высокие дозы препаратов, нередко с включением бензодиазепинов и гормонов. После 9-и летнего возраста частота приступов постепенно снижается; после 11 лет постепенно блокируется эпилептиформная активность; и к началу пубертатного периода пациенты достигают полной электро – клинической ремиссии [Мухин К.Ю. и соавт., 2001; Fejerman и соавт., 2000]. Выраженность когнитивных нарушений также уменьшается с возрастом. Однако у ряда больных когнитивные и речевые расстройства остаются, не смотря на прекращение приступов, что существенно снижает их способность к обучению и социальную адаптацию.
Страницы: [1] 2 Все Вниз
Доброкачественную роландическую эпилепсию относят к генетическим неврологическим заболеваниям. У пациентов отмечается повышенная возбудимость височной коры больших полушарий, что обуславливает возникновение симптомов. Основные клинические проявления — судорожные приступы, имеющие локальный или генерализованный характер. Для подтверждения диагноза необходимо проведение ЭЭГ. Как правило болезнь самостоятельно проходит у детей при взрослении.
Этиология заболевания

Причина развития болезни неизвестна. У 40-70% пациентов отмечается наследование патологии. Оно носит мозаичный характер, т. е. встречается не у всех родственников. На сегодняшний день в неврологии считают, что за развитие роландической эпилепсии ответственны два гена, определяющих работу нейронов коры больших полушарий.
Основная теория объясняет возникновение болезни незрелостью корковых отделов головного мозга, преимущественно центральной части височной доли. Эпилепсия связана с возрастом пациента и возникает преимущественно у детей. Кора больших полушарий в детском возрасте отличается рядом особенностей:
- большое количество возбуждающих нейромедиаторов, превосходящих по количеству тормозящие молекулы;
- µ-аминомасляная кислота и рецепторы к ней выявляются в небольшом количестве;
- большая часть синапсов носит возбуждающий характер;
- эпилептогенные области ЦНС (гиппокамп, лимбическая система) имеют высокий уровень активности.
Указанные факторы по мере взросления ребенка устраняются, что приводит к снижению частоты приступов роландической эпилепсии и полному выздоровлению. У взрослых заболевание не встречается.
Клинические проявления
Парциальные приступы без утраты сознания — основной симптом роландической эпилепсии при ее классическом течении. У больных до возникновения эпиприступа отмечается сенсорная аура: чувство покалывания или онемения в области лица, губ, языка или глотки. После этого появляется моторный компонент в виде тонических, клонических судорог или их сочетания.
Приступы роландической эпилепсии бывают двух форм: фарингооральный и гемифациальный.
- При фарингооральном судороги возникают в мышечных группах глотки, гортани, языка и губ. Они носят односторонний характер и приводят к изменению речи, а также к повышенному слюноотделению. Во время эпиприступа ребенок может издавать неприятные звуки.
- Гемифациальный сопровождается односторонним сокращением мимической и жевательной мускулатуры.
У большинства детей приступы эпилепсии возникают перед сном или сразу после пробуждения. Специалисты считают, что это связано с особенностями работы головного мозга в ночное время. Припадки днем встречаются редко. У 15-25% детей возможно распространение судорог на руку, а у 5% — на ногу.
На фоне парциальных эпиприступов возможно развитие вторично-генерализованных судорожных припадков, которые сопровождаются тоническими и клоническими сокращениями мускулатуры всего тела и потерей сознания.
При атипичной роландической эпилепсии возникают полиморфные приступы. Они могут носить характер фокальных моторных, атонических припадков, атипичных абсансов, генерализованных судорог и эпилептического статуса.
Диагностические мероприятия
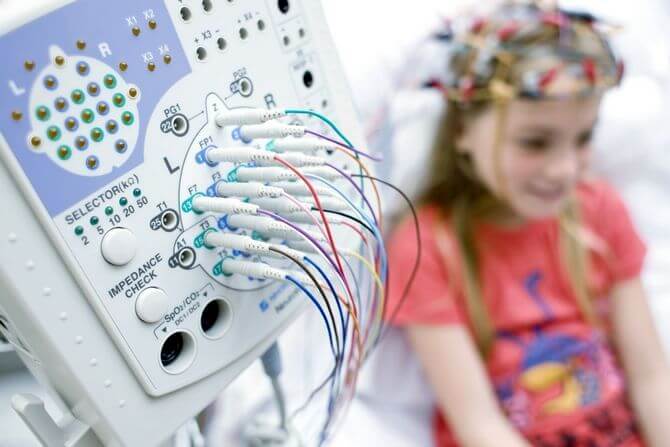
Обследование больных проводится по определенному алгоритму:
Отличия от других заболеваний
При проведении дифференциальной диагностики необходимо исключить органические поражения головного мозга, ночную эпилепсию и синдром псевдо-Леннокса.
Внутримозговые опухоли, изменения при черепно-мозговых травмах, менингите и энцефалите могут привести к появлению эпилептических приступов. Для подобных заболеваний характерно наличие стойкой неврологической симптоматики (снижение мышечной силы, появление парастезий, нарушения зрения и др.). При роландической эпилепсии на ЭЭГ всегда присутствует нормальная основная активность.
У 3-7% больных возможно развитие синдрома псевдо-Леннокса. В этом случае помимо классических приступов возникают абсансы, нарушения памяти и мышления и другие симптомы, что приводит к трудностям в диагностике. При синдроме псевдо-Леннокса при проведении электроэнцефалографии возникают медленные комплексы или диффузная пиковая активность.
Ночная эпилепсия со сложными парциальными приступами — патология, сопровождающаяся схожими симптомами в ночное время. Основное отличие связано с тем, что больные теряют сознание, а на ЭЭГ имеются изменения в активности лобных отделов головного мозга.
Интерпретировать результаты проводимых исследований должен только врач. Попытки самостоятельной постановки диагноза, а также изменение назначенного специалистом лечения может стать причиной прогрессирования патологии или развития побочных эффектов от применения медикаментов.
Подходы к лечению

Однозначного ответа о том, лечить или нет роландическую эпилепсию нет. Патология является доброкачественной и проходит самостоятельно без использования каких-либо медикаментозных средств. Исходя из этого, ряд специалистов отказывается от назначения противоэпилептических препаратов.
Большая часть неврологов считает, что использование лекарственных средств все же необходимо. У больных есть риск развития атипичной фокальной эпилепсии, характеризующейся частыми приступами без тенденции к самостоятельному выздоровлению. Кроме того, под диагнозом роландических эпиприступов могут скрываться другие типы эпилепсии в случае неправильной диагностики. В отсутствии противоэпилептических препаратов, данные патологии способны быстро прогрессировать.
В лечении используют медикаменты на основе вальпроевой кислоты — Депакин, Энкорат хроно и др. Их применяют в виде монотерапии, т. е. не комбинируют с другими средствами. Если вальпроевая кислота не эффективна, то ее заменяют на Леветирацетам. При возрасте ребенка старше 7 лет возможно использование Карбамазепина.
Помимо указанных лекарственных препаратов, специалисты не рекомендуют использовать другие медикаменты с противосудорожной активностью или обладающие психостимулирующим действием. Это может стать причиной развития побочных эффектов, затрудняющих терапию заболевания.
Современные научные исследования показывают, что большое значение в профилактике эпилепсии имеет питание. Положительный эффект наблюдается при переходе на кетогенную диету. Рацион подбирается индивидуально, в зависимости от возраста ребенка, его веса, а также уровня физической и интеллектуальной нагрузки в течение дня. Питание при кетодиете основывается на повышенном потреблении жиров, которые в организме трансформируются в кетоновые тела, улучшающие работу головного мозга. Следует отметить, что во время перехода на диету необходимо постоянно находится под присмотром врача. При этом больному ежедневно определяют уровень кетоновых тел в крови, оценивая эффективность диеты.
Правила первой помощи
Эпилептические приступы у детей возникают в вечернее или утреннее время, так как связаны со сном. Родителям необходимо знать, как оказать первую помощь.
При появлении признаков ауры – слабости, ощущение больным неприятного запаха, его следует уложить на мягкую кровать. Это позволяет предупредить травмы при развитии вторично-генерализованного приступа.
Во время эпиприпадка маленького ребенка нужно приподнять, крепко удерживая его за тело и голову. Это необходимо для предупреждения развития травм в результате ударов о твердые предметы: стену, элементы мебели и др. Ни в коем случае не стоит пытаться удержать больного крепко, сжав ноги и руки. В этом случае имеется риск травматизации мышц и связочного аппарата.
Вызвать скорую медицинскую помощь нужно в том случае, когда приступ носит генерализованный характер и сопровождается потерей сознания.
Предупреждение заболевания

Первичная профилактика патологии отсутствует.
Для профилактики судорожных приступов выделяют ряд рекомендаций:
- Обеспечить ребенку постоянный режим дня с достаточным временем на отдых. Одна из причин развития эпиприпадка — нарушение сна в виде его малой продолжительности.
- Спать больной должен в общей с родителями комнате, так как его нежелательно оставлять одного. Если это невозможно, следует использовать радионяню. Родители, находящиеся рядом с ребенком, могут своевременно заметить ауру и оказать необходимую первую помощь.
- За 1-2 часа до сна необходимо исключить игры за компьютером, смартфоном, а также любую нагрузку, стимулирующую работу головного мозга. Кроме того, не следует плотно кормить детей перед сном.
Прогноз
Прогноз при роландической эпилепсии благоприятный. В возрасте 15 лет у 90% больных наблюдается полное выздоровление без каких-либо остаточных симптомов. У 1-3% подростков эпизодические эпилептические приступы сохраняются, однако, их число не увеличивается.
Неблагоприятный прогноз с прогрессированием патологии возможен в тех случаях, когда первые эпизоды болезни возникают до 4 или после 10 лет жизни. При этом у детей имеется склонность к вторично-генерализованным эпиприпадкам, сопровождающимися потерей сознания. Эффективность медикаментов при этом снижена.
Рецидивы возможны спустя несколько лет бессимптомного периода. Если заболевание сохраняется во взрослом возрасте, то приступы носят единичный характер. Больной может не придавать им значение, так как они возникают в ночное время и могут оставаться незамеченными.
Медицинское наблюдение и применение противоэпилептических средств позволяет предупредить прогрессирование болезни и развитие осложнений. При первых симптомах эпилепсии следует обращаться за профессиональной медицинской помощью к врачу.
Статья по теме: Эпилепсия у детей
Страницы: [1] 2 Все Вниз
Читайте также:
