
Агенезии центральной нервной системы
Агенезия и дисгенезия мозолистого тела Может быть полной или частичной. В последних случаях задняя часть обычно утрачена, из-за того, что мозолистое тело развивается в переднезаднем направлении, тем не менее, возможна и передняя агенезия (Aicardi et al., 1987; Barkovich и Norman, 1988b; Sztriha, 2005). Встречаются атипические формы, сложно отличимые от голопрозэнцефалии (Barkovich, 1990). Агенезия мозолистого тела относительно распространена. Распространенность среди населения в целом оценивается как 3-7/1000 (Bedeschi et al., 2006).
Наблюдаемая частота возросла с введением в практику КТ и MPT. Jeret et al. (1987) выявили 33 случая в серии 1447 КТ снимков.
Даже теперь диагноз в основном ставится только пациентам с неврологическими симптомами, поэтому истинная частота не известна.
Отсутствие мозолистой спайки, как правило, замещается двумя продольными связками, известными как продольное мозолистое тело или пучки Пробста, проходящие по внутренней стороне полушарий. Часто появляются борозды на внутренней стороне с радиальным расположением и расширением затылочных рогов, сохраняющих их фетальную морфологию, так называемую кольпоцефалию (Noorani et al., 1988). Другие связанные аномалии часто включают в себя формирование кист кзади от третьего желудочка, сообщающиеся или несообщающиеся (Yokota et al., 1984, Griebel et al., 1995, Barkovich et al., 2001b) аномалии червя мозжечка и ствола мозга, а также смешанные мальформации ЦНС, такие как гетеротопия, аномалии образования извилин или цефалоцеле (Jeret et al., 1987; Barkovich и Norman, 1988b; Serur et al., 1988).
Гигантские кисты могут иметь благоприятный исход, несмотря на внушительные размеры (Lena et al., 1995; Haverkamp et al, 2002). Пороки развития ЦНС были обнаружены у 33% пациентов с полной и у 42% из них с частичной агенезией (Bedeschi et al., 2006).
Именно эти сочетанные аномалии отвечают за клинические проявления. Липома мозолистого тела почти неизменно сопровождает агенезию этой структуры (Zee et al., 1981; Vade и Horowitz, 1992). Характерны периферические мальформации (Parrish et al., 1979). Особенно часто встречаются глазные аномалии (Aicardi et al., 1987). Гипоплазия мозолистого тела (Bodensteiner et al., 1994) может быть минимальной формой каллозной дисгенезии, но гораздо чаще следствием кортикальных нейрональных потерь.
Этиология разнообразна. Идентифицировано не менее 46 синдромных пороков развития или метаболических расстройств и 30 мутантных генов (Kamnasaran, 2005). При несиндромных формах, генетическая передача встречается редко, хотя имеются сообщения о семейных случаях с аутосомно-рецессивным (Finlay et al., 2000), Х-сцепленным рецессивным (Menkes et al., 1964; Kaplan, 1983) и аутосомно-доминантным типом наследования (Aicardi et al. 1987). Выявлено множество хромосомных дефектов, включая трисомию 8, 13, 16 и 18, а также смесь менее распространенных хромосомных дефектов.
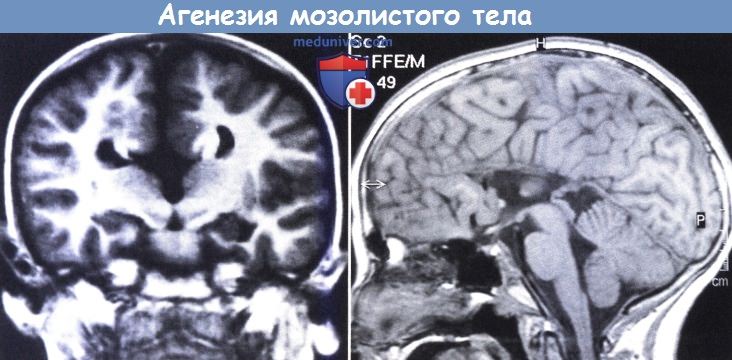
Агенезия мозолистого тела, (слева) МРТ (инверсия-восстановление):
продольное мозолистое тело (пучки Пробста) около внутренней поверхности тел желудочков мозга.
(справа) Сагиттальная проекция: полное отсутствие мозолистого тела и радиальное распределение борозд на внутренней стороне полушарий.
Serur et al. (1988) просмотрели 81 случай из литературных источников, из которых в 21 была трисомия 8, у 14 была трисомия 13-15 и у 18 затронуты хромосомы 17 или 18. Из 34 выполненных кариотипов в двух была обнаружена трисомия 8. Субтеломерные абберации были выявлены в 5% случаев Bedeschi et al. (2006). Среди экологических факторов выделяют плодный алкогольный синдром. Некоторые метаболические заболевания, особенно гипергликемия (Dobyns, 1989), недостаточность пируватдегидрогеназы (Bamforth et al., 1988, Raoul et al., 2003) и другие метаболические расстройства вместе составляют около 2% случаев агенезии мозолистого тела. В большинстве случаев их происхождение неизвестно.
Описание клинических проявлений каллозной аге-незии можно разделить на две части: несиндромные и синдромные формы (Davila-Guttierez, 2002).
Несиндромные формы наиболее распространены (Jeret et al., 1987; Serur et al., 1988). Неизвестный процент случаев остается бессимптомным или случайно выявляется только благодаря большим размерам головы. У большинства пациентов отмечается задержка умственного развития, судороги и/или большие размеры головы (Aicardi et al., 1987). Часто обнаруживаются гипертелоризм. В исследовании Jeret et al. (1987) 82% пациентов имели умственную отсталость или задержку развития, 43% страдали судорогами и у 31% развился церебральный паралич.
Специфические расстройства межполушарной передачи либо отсутствуют, либо только минимальные (Jeeves и Temple, 1987).
Тем не менее, имеются сообщения о тонких нарушениях межполушарной связи и топографической памяти. В редких случаях может наблюдаться эндокринологическая патология (Paul et al., 2003).
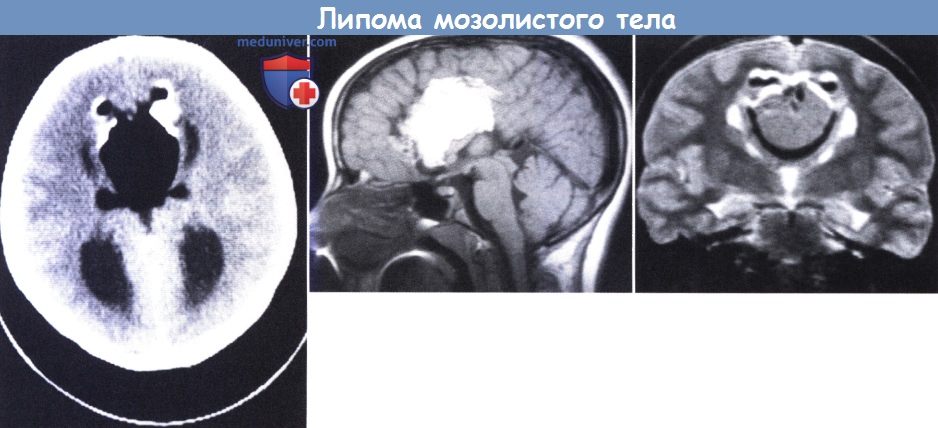
Липома мозолистого тела у 8-летней девочки с парциальным комплексом судорог, но без неврологических нарушений.
(слева) КТ: крупные массы жировой плотности, разделенные передними рогами боковых желудочков, с периферической кальцификацией и двумя небольшими латеральными участками распространения жировых масс.
(в центре) МРТ (сагиттальная проекция): замещение мозолистого тела тканью липомы.
(справа) МРТ (Т 2-взвешенная последовательность): полное замещение каллезного тела жировой тканью. 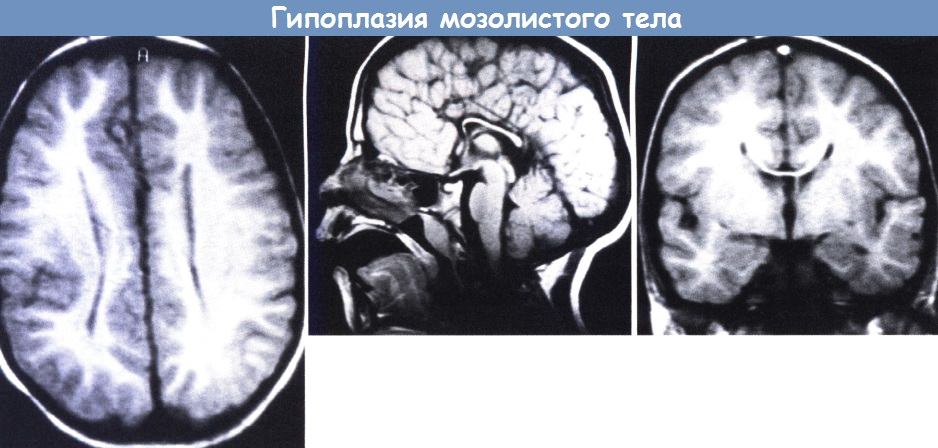
Гипоплазия мозолистого тела, (слева) МРТ (аксиальная проекция): вид желудочков в соответствии с агенезией мозолистого тела.
(в центре) Сагиттальная проекция: полное мозолистое тело с коленом и валиком, но укороченное и истонченное. Обратите внимание на радиальное расположение медиальной извилины.
(справа) Фронтальная проекция: широкое разделение тел желудочков пролабирующей лимбической извилиной.
Синдромные формы перечислены в таблице ниже.
Синдром Айкарди (Chevrie и Aicardi, 1986, Aicardi, 2005) имеет отношение примерно к 1% случаев с инфантильными спазмами, вероятно, из-за Х-сцепленных доминантных мутаций. Встречается почти исключительно у девочек, хотя имеется сообщение о двух случаях у мальчиков с XXY набором хромосом. Известно только об одном семейном случае у двух сестер (Molina et al., 1989). Характерные особенности синдрома включают в себя инфантильные спазмы и специфические хориоидальные лакуны, часто в сочетании с колобомой зрительного диска. Позвоночно-реберные аномалии имеются в половине случаев. Исход обычно неблагоприятный, с сохраняющимися судорогами и глубокой умственной отсталостью. Спектр тяжести оказался шире, чем предполагалось ранее (Menezes et al, 1994). В редких случаях может присутствовать мозолистое тело (Aicardi, 1994, 1996).
Диагноз определяют хориоидальные лакуны и сопутствующие аномалии, выявленные при МРТ (перивентрикулярная гетеротопия, диспластичная кора, эпендимальные кисты). При патологическом исследовании в мозге обнаруживают многочисленные участки гетеротопии и полимикрогирии не разделенного на слои типа (Billette de Villemeur et al., 1992), тогда как так называемые лакуны представляют собой истончение пигментного эпителиального и сосудистого слоя с утратой пигментных гранул. Эпендимальные кисты часто обнаруживают вокруг третьего желудочка. Кисты или опухоли сосудистого сплетения могут достигать больших размеров (Aicardi, 2005). При выявлении вместе с агенезией мозолистого тела возможен пренатальный диагноз.
Другие синдромные формы встречаются редко или в большинстве ограничены определенными этническими группами.

Синдром Айкарди у трехмесячной девочки.
Обратите внимание на асимметрию полушарий, двусторонние кисты сосудистого сплетения, кисты вокруг третьего желудочка с различным сигналом от цереброспинальной жидкости и перивентрикулярные гетеротопии.
Эпендимальное происхождение кист сосудистого сплетения было подтверждено при гистологическом исследовании. 
Агенезия мозолистого тепа. Ультразвуковое антенатальное исследование, сагиттальная проекция.
Обратите внимание на нормальный четвертый желудочек, отсутствие эхосигнала от мозолистого тела и расширенный боковой желудочек.
Слева на фотографии — затылок плода.
Семейный синдром агенезии мозолистого тела с патологией гениталий, который также может проявляться с микроцефалией и другими аномалиями ЦНС, является частью более обширного спектра расстройств, связанных с мутациями в гене ARX на хромосоме Хр22.3 (Hartmann et al, 2004).
Синдром Андерманна был описан у французского канадца в районе озера Сент-Джонс (Andermann, 1981), но о нескольких случаях было заявлено за пределами Канады. Этот синдром затрагивает периферическую нервную систему в дополнение к агенезии мозолистого тела или гипотрофии. Агенезия мозолистого тела зачастую является частью рото-лице-пальцевого синдрома I типа.
Пренатальная диагностика возможна с 22 недели (Bennett et al., 1996; Simon et al., 2000a). Решение о прерывании беременности сложно принять безоговорочно, пока нет данных о распространенности бессимптомных случаев. Blum et al. (1990) сообщали, что у 6 из 12 новорожденных, у которых агенезия мозолистого тела была диагностирована антенатально, имели нормальное развитие в возрасте 2-8 лет. Moutard et al. (2003) наблюдали 17 случаев с пренатально диагностированной изолированной агенезией с повторными измерениями IQ. В возрасте 6 лет все дети имели коэффициент умственного развития на нормальном уровне с тенденцией к нижней границе нормы. Девять детей в исследовании Bedeschi et al. (2006) имели нормальное развитие.
Тем не менее, у 2 из 9 детей, пренатально диагностированных с помощью МРТ без сочетанных аномалий, отмечалась задержка в развитии (Volpe et al., 2006). Случаи, связанные с другими мальформациями или хромосомными аномалиями, неизменно имели неблагоприятный исход. Поэтому крайне важно фетальное кариотипирование и полное обследование на наличие сочетанных пороков развития.

Редактор: Искандер Милевски. Дата публикации: 30.11.2018
Агенезия мозолистого тела (по Mingazzini).
по виду напоминают эмбриональные невро-бласты; отсутствует нормальная диферен-циация корковых клеток, отчего становится незаметным послойное строение коры; нередко заметно преобладание полиморфного слоя. Ганглиозные клетки бедны прото-плазматическими отростками; ядро богато хроматином; тигроидного вещества мало. Невроглия представляется нормальной или относительно гипертрофированной. Корковых миэлиновых волокон мало; они тонки и бедны миэлином; преимущественно поражаются ассоциационные волокна, в особенности тангенциальная система, в меньшей степени—суперрадиальные и интеррадиальные волокна; проекционные системы кажутся лучше развитыми. Корковые А. являются обычной находкой при изучении головного мозга б-ных врожденным слабоумием. Помимо кортикальных А. с их различной локализацией, встречаются также А. коммиссуральных систем; из них особого внимания заслуживает агенезия мозолистого тела, которая бывает как полной, так и частичной; в последнем случае иногда можно встретить только колено или утолщение; при полной А. отсутствуют также striae Lancisii; taeniae tectae иногда сохраняются (см. рисунок). При полной А. мозолистого тела наблюдаются психические расстройства; по Мингацини (Mingazzini), из 56 случаев А. мозолистого тела (при чем в 40 случаях была полная А.) в 38 случаях было психическое расстройство (чаще всего идиотия). Описаны А. передней спайки, прозрачной перепонки, перекреста зрительных нервов, зрительного канатика, шишковидной железы и т. п. В стволовой части описаны А. ядер отводящего, лицевого и подъязычного нервов.—А. мозжечка чаще всего бывает частичной; полная А., по Тома (Thomas), встречается чрезвычайно редко. В случаях полной А. мозжечка мозк-но все-таки найти части vermis и flocculus; остальная масса мозжечка замещается утолщенной мягкой мозговой оболочкой; при частичной А.мозжечка, когда недоразвивается одно из его полушарий, мягкая оболочка также бывает утолщенной; при исследовании недоразвитого полушария можно констатировать отсутствие коркового слоя при сохранности nucl. fastigii и nucl. dentati; иногда на стороне недоразвитого полушария отсутствует сосудистое сплетение четвертого желудочка (Brun, Edinger). Симптомы агенезии мозжечка обыкновенно обнаруживаются уже в самом раннем детском возрасте, когда ребенок начинает ходить; он часто падает, наблюдаются дрожание рук, неловкость движений, расстройство артикуляции, замедленность речи.—При частичной А. спинного моз-г а, б. ч. зависящей от задержки развития большого мозга и мозжечка, спинной мозг представляется уменьшенным в своих поперечных размерах; на дорзальной стороне боковых столбов встречается глубоко врезывающаяся бороздка; количество серого вещества передних рогов уменьшено; со стороны проводников отмечается недоразвитие церебро- и церебелло-сшшальных путей.— Патогенез различных А. не вполне выяснен. При вскрытии больных с А. ц. н. с. много раз были найдены пат.-гистолог. изменения желез внутренней секреции, напр., надпочечников, зобной железы, щитовидной, мозгового придатка; существование агенезии многие исследователи ставят в причинную связь с нарушением функций эндокринных желез. Лит.: Ernst, Die Missbildungen des Nerven-systems, 1909; Brun, Das Kleinhim, 1927; Nou-veau traite de medecine, publ. par Roger G., Wi j d a 1 F.et Teissier P., rase. 19, Paris, 1925: a) L e-vi-Valensi, Eneephalopathies infantiles, b) Andre-Thomas, Pathologie du cervelet; Handbuch der pathologischen Anatomie des Nervensystems; Flatau F., Jacobsohn L. u. Minor L., 1903: a) Anton, Entwicklungsstorungen des Ge-hirns, b) Petren, Die Entwicklungsanomalien des Riickenmarks; Mingazzini G., Der Balken, В., 1922. А. Капустин.
Большая медицинская энциклопедия . 1970 .
- АГЕВЗИЯ
- АГЕНЕЗИЯ
ГОЛОВНОЙ МОЗГ — ГОЛОВНОЙ МОЗГ. Содержание: Методы изучения головного мозга . . . 485 Филогенетическое и онтогенетическое развитие головного мозга. 489 Bee головного мозга. 502 Анатомия головного мозга Макроскопическое и… … Большая медицинская энциклопедия
МОЗЖЕЧКОВО-МОСТОВОЙ УГОЛ — (Klein hirnbruckenwinkel, angle ponto cerebelleuse, по нек рым angle ponto bulbo cerebelleuse) занимает своеобразное место в невропатологии, неврогистопатологии и неврохирургии. Названием этим обозначается угол между мозжечком, продолговатым… … Большая медицинская энциклопедия
Головно́й мозг — (encephalon) передний отдел центральной нервной системы, расположенный в полости черепа. Эмбриология и анатомия У четырехнедельного эмбриона человека в головной части нервной трубки появляются 3 первичных мозговых пузырька передний… … Медицинская энциклопедия
НЕРВНАЯ СИСТЕМА — НЕРВНАЯ СИСТЕМА. Содержание: I. Эмбриогенез, гистогенез и филогенез Н.с. . 518 II. Анатомия Н. с. 524 III. Физиология Н. с. 525 IV. Патология Н.с. 54? I. Эмбриогенез, гистогенез и филогенез Н. е.… … Большая медицинская энциклопедия
ОРГАН — (от греч. organon орудие), определенная совокупность нескольких тканей, обладающая особой функцией. Клетки животного организма, группируясь, образуют анат. единицы более высокого порядка ткани. Последние, соединяясь, в свою очередь дают анат.… … Большая медицинская энциклопедия
УРОДСТВА — УРОДСТВА, врожденные стойкие нарушения взаимоотношений отдельных частей организма, возникающие в течение индивидуального развития и выходящие за пределы вариаций этих взаимоотношений у данного вида. У. может касаться строения всего организма в… … Большая медицинская энциклопедия
НАДПОЧЕЧНИКИ — (glandulae suprarena les, epinephra, hypernephra, paraganglia), парные инкреторные органы, лежащие в задне верхней части брюшной полости на верхневнутренней поверхности почек. Открытие и первое описание надпочечников приписывается анатому… … Большая медицинская энциклопедия

Перинатальное поражение центральной нервной системы (МКБ-10 – G00-G99) и патоморфологические состояния головного мозга новорожденных являются результатом вмешательства патогенного агента в текущий процесс развития поврежденной области. Окончательной морфологии способствуют реактивные и репаративные изменения в сочетании с продолжением гистогенетического развития.
Важность детальных знаний о внутриутробных и перинатальных повреждениях головного мозга и ППЦНС у детей значительно возросла в последние годы в связи с развитием интенсивной терапии патологических новорожденных, пренатальной ультразвуковой диагностики. Диагноз ППЦНС у ребенка – это осложняющий фактор его дальнейшего развития, составляющий важную часть педиатрии и неврологии. Патологии ЦНС – это фактор риска ряда других болезней, в частности, рассеянного склероза.
Гипоксическая ишемическая энцефалопатия новорожденных (ГИЭ)
ГИЭ – это наиболее распространенное патологическое обнаружение в мозге новорожденных. Заболевание происходит внутриутробно, внутриматочно и послеродово. Оно вносит значительный вклад в процент смертей новорожденных, в различные формы двигательного и психологического нарушения.
Гипоксическая ишемическая энцефалопатия связана с целым рядом причин, среди которых:
- преждевременное отделение плаценты;
- компрессия пуповины;
- непрогрессирующие роды;
- травма ЦНС при родах;
- аномалии верхних дыхательных путей;
- аспирация околоплодных вод;
- пневмоторакс;
- врожденная диафрагмальная грыжа;
- трахео-эзофагеальная фистула;
- пневмония надпочечников и другие факторы.
Повреждение коры при ГИЭ в основном отмечается у зрелых новорожденных. Зрелая кора требовательна к снабжению кислородом. Типичный для расстройства ламинарный некроз III-V слоя, где нейроны имеют высокую метаболическую активность. Очаговый некроз распространен в области деления нижних желобков мозга. Полный некроз поражает кору по всей высоте. После некроза возникает репаративный астроцитоз, извилины сужаются, становятся нерегулярными (улегирия).

Наиболее типичное состояние – перивентрикулярная лейкомаляция. Это небольшие транзиторные инфаркты, расположенные вокруг боковых желудочков примерно в 0,5 см от стенки. Они находятся в зоне деления между центростремительными и центробежными артериями белого вещества. Редко располагаются вокруг межжелудочкового отверстия и затылочного угла боковой камеры. Инфаркты могут быть множественными, распределяться по большей части белого вещества полушарий.
Небольшой некроз излечивается глиальным рубцом, после объемного остаются псевдокисты.
Некроз – это тяжелая форма гипоксического повреждения (градус III). При легкой гипоксии избирательно затрагивается часть глии и меньшее количество аксонов, заживление приводит к гипоплазии белого вещества. Легкая гипоксия вызывает только отек белого вещества (градус I), обратимое изменение.
Повреждение базальных ганглиев (таламус, стриатум, бледный шар) в основном сопутствует повреждению коры. Некроз залечивается астроглиальным рубцом с атипичными процессами миелинизации нервных волокон и астроглии. Макроскопически поврежденные участки уменьшены, нерегулярно мраморизованы.
Повреждение ствола головного мозга, мозжечка и спинного мозга сопровождается серьезными изменениями в коре и подкожной клетчатке. Избирательно гипоксическое повреждение возникает после одного случая асфиксии повышенной тяжести, связанной с кратковременной остановкой сердца.
В части нейронов в мосте, в продолговатой части, базальном ядре и в ядрах четверохолмия происходит регрессия и некроз. Если новорожденный выживает, следует астроглиоз, ствол мозга уменьшается, становится жестким. Ишемическое повреждение нейронов спинного мозга связано с остановкой сердца. В мозжечке зернистый слой более чувствителен к гипоксии, чем слой клеток Пуркинье.
В генезе некроза имеется общая гипоксия, сдавливание позвоночных артерий при экстремальном положении головы во время родов.
Понтосубикулярные некрозы косвенно связаны с ГИЭ. Они вызваны реализацией терапевтической гипероксии при лечении гипоксических состояний. Это избирательные некрозы нейронов моста, базальных ядер и субикулума. После заживления развивается глиоз.

Очаговые и многоочаговые гипоксически-ишемические изменения возникают при блокировке отдельных мозговых артерий. В соответствующей артерии происходит инфарктоподобный некроз с последующим образованием псевдокисты. Степень изменений зависит от размера заблокированных сосудов (от небольшого очага в 1-5 мм до возможного поражения всей доли).
Причины блокировки бывают пренатальными и постнатальными, включающими:
- изоиммунную тромбоцитопению;
- эмболию плаценты;
- тромбоэмболию из закрывающегося артериального протока;
- артериальную диссеминированную внутрисосудистую коагуляцию;
- тромбоз при лептоменингите, характерном, как для раннего постнатального периода, так и для более позднего детского возраста (грудничков).
Дальнейшее развитие выживших детей, последствия перинатального поражения ЦНС зависят от степени и расположения острого повреждения. После заживления острой фазы более поздние хронические состояния проявляются психомоторной отсталостью. С клиническими данными коррелируют следующие морфологические состояния:
- Минимальные поражения головного мозга. Они не имеют достоверно определенной морфологии. При непрерывном исследовании головного мозга в коре можно обнаружить небольшие глиальные шрамы или очаговые нарушения, крошечные астроглиальные шрамы в белом веществе в количестве до 25%.
- Улегирии. Изначально представляют собой нормально сформированные извилины, вторично нерегулярно загибаемые и сужаемые. Изменение может быть отмечено на отдельных извилинах, но также нарушение может затронуть большую часть поверхности полушария. Улегирия распространена в области парасагитального деления. Нейроны отсутствуют, извилины превращаются в глиофиброзный рубец.
- Гидранэнцефалия. Одно или оба полушария замещаются тонким глиомезенхимальным слоем. Патология возникает после обширного разрушения незрелых полушарий мозга.
- Гипоплазия белого вещества. Представляет собой диффузное уменьшение объема белого вещества. Возникает после полной длительной гипоксии недоношенных новорожденных из-за множественного очагового некроза и диффузного повреждения незрелой глии.
- Псевдокисты. Возникают после заживления более крупных некрозов; одиночные являются последствием окклюзии одной артерии, множественные – результатом многоочаговой кистозной энцефалопатии. Полушария, иногда и базальные ганглии, пронизаны многочисленными нерегулярными, часто взаимно связывающимися клетками, разделенными щелями глиофиброзной ткани. Нейроны исчезают. Осложнение является результатом тяжелой диффузной гипоксии или множественной окклюзии сосудов.
Дефекты развития ЦНС
На мальформации приходится 19% причин перинатальной смертности, из которых за 50-70% отвечают дефекты развития ЦНС, представляющие непосредственную угрозу жизни.
Мальформация может иметь морфологическое проявление только в ЦНС; но часто одновременно поражаются другие системы (кожа, кости, мышцы, паренхиматозные органы).
Развитие ЦНС можно разделить на 5 периодов:
- Период дорсальной индукции (3-4 недели внутриматочного развития).
- Период вентральной индукции (5-6 неделя внутриутробного развития).
- Нейробластический период – глиобластная пролиферация (2-6-месячный внутриматочный период).
- Период локальной дифференцировки клеток (с 5-го внутриутробного месяца, продолжение в постнатальном периоде).
- Период миелинизации (с 7-го внутриутробного месяца до постнатального периода).
Большинство обнаружений можно надежно отнести к отдельным периодическим группам.
К этой группе перинатальных поражений ЦНС у новорожденных относятся следующие расстройства:
- Тотальный краниосхиз. Это полный дефект закрытия нервной трубки. Нейрокраниум отсутствует, позвоночный канал частично или полностью расщеплен. Мозга нет (анэнцефалия), лицевая часть навязывает прямо на укороченную грудь. Глаза обращены краниально (ураноскопия), основание черепа покрыто сосудистой мембраной (область нейроаскулозы), проходящей периферически в коже.
- Энцефалоцеле. Нейрокраниальный дефект, связанный с пролапсом оболочек и мозга. Макроскопически он имеет вид сферической формы диаметром несколько сантиметров, покрыт кожей на поверхности. Из-за дефекта прогибается жесткая оболочка и часть мозга, прогиб только жесткой оболочки встречается реже.
- Рахишизис. Это дефект закрытия позвоночного канала. Расстройство часто связано с краниосхизом. Спинная расщелина может быть полной или ограниченной.
- Порок развития мозжечка. Это заболевание в основном является частью более сложных пороков развития мозга. Агенезия встречается редко. Гипоплазия может поражать весь мозг или его часть (черви мозжечка, полушария).
Эта группа ППЦНС у новорожденных включает такие патологии, как голопрозэнцефалия и менее выраженный лицевой дисморфизм.

Это дефект с характерной комбинацией аномалий головного мозга и головы. Это хромосомный дефект, чаще встречающийся у диабетических матерей. Руководящей основной диагноза являются лицевые аномалии:
- циклопия – наличие только одной орбиты и одного глазного яблока, отсутствие носовой полости;
- орбиты, расположенные близко друг к другу, имеют этмоцефалию (гипотелоризм), в небольшой носовой полости отсутствует перегородка;
- премаксилярная агенезия характеризуется гипотелоризмом, плоским носом, средним расщеплением верхней губы и верхней челюсти.
Это смешанные аномалии, такие как гипотелоризм, гипертелоризм, очень плоский нос, односторонние и двусторонние расщелины лица.
Группа пролиферативных нарушений включает дефекты, вызванные уменьшенной, чрезмерной или нетипичной пролиферацией зародышевых слоев мозга, незрелого белого или серого вещества. В эту группу входят первичные изменения размера мозга (макроцефалия и микроцефалия), состояния, характеризующиеся чрезмерной пролиферацией некоторых компонентов ткани (нейрокутанные синдромы).
Мозг уменьшается по сравнению с нормой (менее 2 стандартных отклонений от нормы), но все анатомические компоненты создаются пропорционально. Признаки предшествующего повреждения отсутствуют (послевоспалительные состояния, гипоксия, пороки развития). Предположительное время появления патологии – 2-4-й внутриматочный месяц.
В симптоматике нет значительных изменений моторики, интеллект обычно снижен. В анамнезе течения беременности присутствует фенилкетонурия, применение противоэпилептических препаратов, гиперавитаминоз А, вирусная инфекция, этилизм.
Микроцефалия также является частью ряда хромосомных синдромов.
Это гетерогенная группа с общим признаком – большим мозгом, в котором пропорционально созданы все анатомические компоненты. Клинически регистрируется снижение интеллекта. При церебральном гигантизме макроцефалия связана с общим ускоренным ростом в детстве (большие руки, ноги, челюсть, долихоцефалия, нормальный или слегка сниженный интеллект). В отдельных случаях этиология совершенно неясна.

С 5-го внутриутробного месяца все анатомические части мозга уже четко сформированы. Поражение проявляется нейродинамичными нарушениями, регрессивными изменениями и последующим глиальным рубцеванием. Кроме того, каждое повреждение изменяет процесс миелинизации, развивающийся преимущественно постнатально. Процессу миелинизации препятствуют также метаболические заболевания и воспаления.
Перинатальная травма ЦНС и внутричерепное кровоизлияние
Роды всегда связаны с риском травмы головы и позвоночника у новорожденных. Обычно травмы вызываются диспропорцией между размером головы и шириной родового канала, патологическим положением плода в матке, использованием щипцов и другими факторами. Существует ряд типичных состояний при травмах головы:
- Родовая опухоль. Скопление серозного геморрагического транссудата между сухожильным шлемом и надкостницей. Симптомы включают отек кожи и подкожной клетчатки, диаметром около 6 см, возникающие в месте налегания головы на шейку матки. В патологическом положении плода родовая опухоль может находиться в другом положении. Отек исчезает через 24 часа после родов.
- Кефалогематома. Это субпериостальная гематома над выпуклостью плоских черепных костей. Размер варьируется от 1 см до размера теннисного мяча. Отек не распространяется за край пораженной кости.
- Отступ черепной кости. Это вдавливание гибкой плоской кости черепа новорожденного, диаметром 3-4 см. Внутри кости обычно присутствует трещина.
- Переломы плоской кости имеют форму щелей, идущих радиально и периферически от приподнятой части кости. Часто встречается сочетание с кефалогематомой.
- Остеодиастаз затылочной кости. У новорожденного затылочная кость состоит из 4 частей, связанных синхондрозом. При сдвиге синхондроза может произойти разрыв синусов твердой мозговой оболочки и повреждение ствола мозга.
- Деформация головы. Приблизительно сферическая форма зрелой головки плода в процессе рождения несколько расширяется цилиндрически. В узких родовых путях деформация чрезмерна, края плоских костей движутся друг над другом, повышая риск травмы мозга.
Субарахноидальное кровотечение в форме небольших очагов диаметром 1-10 мм является отдаленным признаком гипоксической энцефалопатии и сепсиса. Объемные субарахноидальные гематомы являются частью крупных желудочковых гематом.
Это частое нахождение у недоношенных новорожденных. Как правило, желудочковая система заполнена свернувшейся кровью, кровоток продолжается через ромбовидное отверстие в субарахноидальное пространство. Чаще всего кровью заполнена только одна боковая камера. Источник гематомы в основном представлен треснувшей субэпендимальной гематомой, депонированной в клеточном перивентрикулярном зародышевом слое.
У доношенных новорожденных желудочковые гематомы исключены.
Эти расстройства всегда являются частью травмы позвоночника. Наиболее распространенное изменение вокруг спинного мозга – эпидуральная гематома. Если она объемная, то может повредить спинной мозг, стать источником затрудненного дыхания новорожденного. Механическое повреждение спинного мозга происходит во время сильного вытяжения при непрогрессивных родах и после использования щипцов.
Лечение
В большинстве случаев для предотвращения серьезных последствий перинатального поражения требуется сердечно-легочная реанимация сразу после рождения и других необходимых действий для обеспечения стабильного состояния и минимизации произошедшего поражения. После успешной реанимации и первичных восстановительных действий у младенца, рожденного после 36-й недели беременности, прекращается нагревание, происходит переход к некоторым из контролируемых методов охлаждения.
Кроме того, необходимо стабилизировать дыхательную активность путем ведения искусственной вентиляции легких. Компенсирование кровяного давления, сердечной недостаточности, регулирование сердечного ритма и капиллярного возврата обеспечивает стабилизацию сердечно-сосудистой системы. В соответствии с лабораторными результатами, измерениями уровня глюкозы в крови, ионограммой стабилизируется внутренняя среда, назначаются соответствующие лекарства (один из используемых препаратов – Кортексин).

Путем выведения жидкости, применения диуретиков достигается противоотечный эффект. При развитии судорожной мышечной активности, под наблюдением невролога назначается противосудорожная терапия (Диазепам, Фенобарбитал) и массажи. Цель такого лечения состоит в достижении полного исчезновения или, как минимум, облегчении судорог.
В зависимости от фактической зрелости и уязвимости структур головного мозга, вследствие гипоксии или ишемии, происходит их повреждение в различной степени. Возбуждающие аминокислоты, активация NMDA-рецепторов с последующим рефлюксом кальция в клетки также играют ключевую роль. При отсутствии энергетической активности нервной клетки это состояние приводит к ее гибели. NMDA-рецептор – это распространенный нейромедиатор (N-метил D-аспартар). Носитель NMDA имеет интересную особенность: он отвечает за создание следов памяти в мозге, особенно в центре памяти (гиппокампе).
В некоторой степени блокировать активацию NMDA-рецептора можно путем применения магния в форме уколов ввиду его нейропротекторного эффекта.
Нет необходимости лечить ППЦНС у новорожденных с использованием всех указанных выше мер. При определении подходящего терапевтического подхода следует учитывать общее состояние ребенка, сроки беременности, на которых он родился, наличие других сопутствующих расстройств. Также необходимо следить за серьезностью гипоксии, ее проявлениями.
Для проведения контролируемой гипотермии существуют точные критерии. Этот метод предназначен исключительно для доношенных детей. Он проводится только в специализированных отделениях интенсивной терапии и реанимации, которые оснащены для этой операции не только технически, но и профессионально подготовленным персоналом.
Прогноз перинатального поражения ЦНС довольно сложен. Существуют определенные взаимосвязи между типичными случаями и дальнейшим развитием заболевания, что, несомненно, влияет на нейромоторное состояние, психическое, эмоциональное и интеллектуальное развитие ребенка. Поэтому необходимо уделять большое внимание надлежащему лечению, дополнять его реабилитационной и педагогической психологической помощью (дизартрия, нарушение когнитивных способностей). После правильной терапии много детей могут жить нормальной жизнью. Но без лечения прогрессирующее заболевание редко не приводит к смерти.
Читайте также:
