
Амилоидоз поражения нервной системы
Классификация амилоидоза
Профилактика амилоидоза
Поражение органов амилоидозом
Симптомы амилоидоза
Поражение почек при амилоидозе
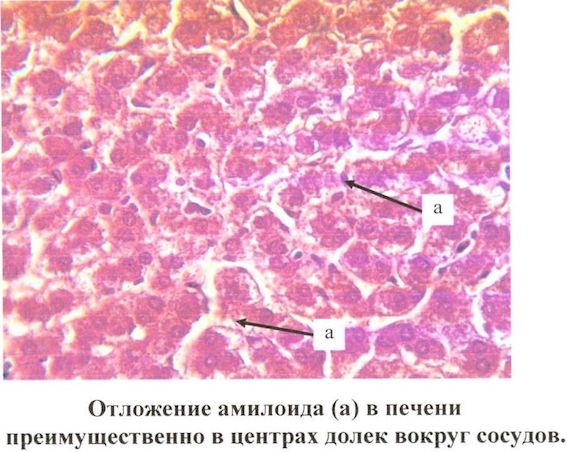
Поражение печени при амилоидозе
Поражение кишечника при амилоидозе
Поражение нервной системы при амилоидозе
- Онемение
- Дизестезии (нарушения чувствительности). Чувствительность при этом страдает больше поверхностная, нежели глубокая
- Жгучие боли в кистях и стопах.
Поражение мышечной ткани при амилоидозе
Поражение кожи при амилоидозе
Диагностика
Виды заболевания
- Первичный или идиопатический. При подобном виде отложения встречаются во всех внутренних органах. Определить точную причину появления невозможно. Наблюдаются множественные аномалии в клетках.
- Вторичный, как осложнение какого-либо заболевания воспалительного характера. Вторичный амилоидоз образуется во внутренних органах. Наблюдается нарушение деятельности органов с наибольшим отложением амилоида – в области почек, селезенки, печени или лимфоузлов. В дальнейшем поражение распространяется на остальные органы с последующим смертельным исходом.
- Наследственный. Подобная форма образуется из-за генетических аномалий в клетках иммунной системы, приводящих к появлению амилоидобластов. Подобная патология диагностируется в некоторых национальных группах или в определенной географической области. К наследственной форме можно отнести:
- Периодическая болезнь – семейная лихорадка Средиземноморья;
- Семейный нефропатический или английский амилоидоз;
- Наследственный нейропатический амилоидоз – португальский, американский или финский;
- Наследственный кардиопатический или датский амилоидоз.
- Старческий. Системный подход позволяет выявить данную патологию у людей после 80 лет.
- Церебральный или мозговой амилоидоз. Диагностируется при болезни Альцгеймера;
- Амилоидоз сердца. Поражает сердечную мышцу. Отложения образуются также в легких, печени и поджелудочной железе.
- Опухолевый. В этом случае амилоидоз развивается локально в органах с выраженным злокачественным процессом. Его причиной становится рак щитовидной железы или опухоль островков поджелудочной железы.
- Гемодиализный. При гемодиализе, назначаемом пациентам с почечной недостаточностью, постепенно повышается содержание в крови B 2 -микроглобулина. Данный белок при взаимодействии с нуклеопротеидами оседает в тканях почек.
Диагностика амилоидоза
- Общий анализ крови. Определяет отклонения от нормы, специфические для амилоидоза. На последних стадиях болезни данное исследование помогает выявить поврежденный орган.
- Общий анализ мочи. Диагностика амилоидоза почек показывает вероятность развития воспалительных процессов, проходящих в почках.
- Протеинурия – содержание в моче белка свыше 3 г/л;
- Гематурия – обнаружение в моче эритроцитов;
- Лейкоцитурия – присутствие в моче лейкоцитов;
- Цилиндрурия – содержание в моче цилиндров, образующихся при амилоидозе из белков, клеток эпителия почек, лейкоцитов и эритроцитов;
- Снижение плотности мочи.
- Биохимический анализ крови. Дает возможность оценить общее состояние организма и установить причину возникновения амилоидоза. При этом анализе определяются:
- Белки общей фазы воспаления, вырабатываемые печенью или определенными лейкоцитами при воспалительном процессе. Особое внимание стоит уделить количеству фибриногена.
- Печеночные пробы указывают на состояние данного органа.
- Увеличение уровня холестерина является признаком нефротического синдрома.
- Снижение уровня белков указывает на нефротический синдром или печеночную недостаточность.
- Повышение количества мочевины и креатинина служит показателем почечной дисфункции при амилоидозе.
Что такое амилоидоз?
Амилоидоз — системное заболевание, которое подразделяется на множество типов и характеризуется поражением паренхиматозных органов (т.е. щитовидной железы, легких, почек, селезенки, печени). Итогом неправильного формирования и избыточного накопления в межклеточном пространстве сложного низкомолекулярного, нерастворимого белка, или так называемого белково-полисахаридного комплекса, служит склероз и атрофии в тканях, и как следствие приводит к недостаточности описанных выше органов.
Данная патология относительно молодая и была выявлена немецким ученым Шлейденом М.Я. в 1983 г., который доказал участие грубодисперсных белков в образовании амилоида.
Признаки и симптомы
Клинические симптомы амилоидоза могут быть разнообразными и зависят от выраженности и локализации амилоидных отложений, биохимического состава амилоида, длительности заболевания, степени нарушения функции органов. Скрытый период амилоидоза, когда отложения гликопротеида могут быть обнаружены только микроскопически, не отличается развитием значимых признаков. По мере прогрессирования функциональной недостаточности пораженного органа нарастают клинические симптомы болезни.
При амилоидозе почек длительно текущая стадия умеренной протеинурии сменяется возникновением нефротического синдрома. Переход к развернутой стадии может быть связан с перенесенной интеркуррентной инфекцией, вакцинацией, переохлаждением, обострением основного заболевания. У больного отмечается постепенное увеличение отеков, возникновение нефрогенной артериальной гипертензии и почечной недостаточности. Иногда развивается тромбоз почечных вен. Массивная потеря белка сопровождается развитием гипопротеинемии, гиперфибриногенемии, гиперлипидемии, азотемии. В моче обнаруживается микро-, иногда макрогематурия, лейкоцитурия.
При амилоидозе сердца отмечается развитие рестриктивной кардиомиопатии с типичными клиническими признаками – кардиомегалией, аритмией, прогрессирующей сердечной недостаточностью. У больного появляется одышка, отеки, слабость, возникающая даже при незначительных физических нагрузках. В редких случаях при амилоидозе сердца возникает полисерозит, проявляющийся возникновением асцита, экссудативного плеврита и перикардита.
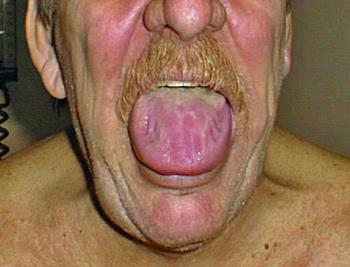
Поражение пищеварительной системы при амилоидозе характеризуется амилоидной инфильтрацией языка, пищевода, желудка, кишечника. Возможно возникновение желудочно-кишечных кровотечений. При амилоидной инфильтрации печени возникает гепатомегалия, холестаз, портальная гипертензия. Поражение поджелудочной железы при данной патологии может маскироваться под хронический панкреатит.
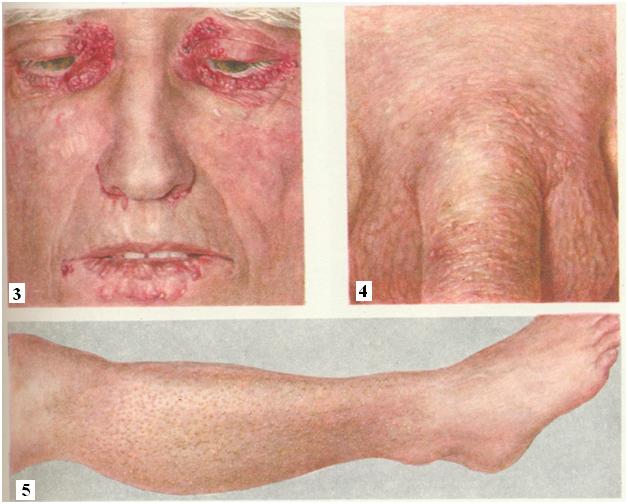
Амилоидоз кожи характеризуется появлением множественных восковидных бляшек в области лица, шеи, естественных кожных складок. По внешним признакам кожные поражения могут напоминать склеродермию, нейродермит или красный плоский лишай.
При поражении суставов типично развитие симметричного полиартрита, запястного туннельного синдрома, плече-лопаточного периартрита, миопатии. Отдельные формы амилоидоза, связанные с поражением нервной системы, могут сопровождаться развитием полинейропатии, параличем нижних конечностей, головными болями, головокружениями, ортостатической гипотензией, потливостью, деменцией.
Причины и факторы риска
Причины развития первичного амилоидоза на данный момент до конца не изучены. При этом установлено, что развитие вторичного амилоидоза обычно связано с хроническими инфекционными (туберкулез, сифилис, актиномикоз) и гнойно-воспалительными заболеваниями (остеомиелит, бронхоэктатическая болезнь, бактериальный эндокардит), реже заболевание связывают с опухолевыми процессами (лимфогранулематозом, лейкозом, раком висцеральных органов).
Развитию реактивной формы амилоидоза подвержены лица, страдающие атеросклерозом, псориазом, ревматическими заболеваниями, например, ревматоидным артритом, болезнью Бехтерева, хроническими воспалительными заболеваниями, такими как неспецифический язвенный колит, болезнь Крона, такими мультисистемными поражениями, как болезнь Уиппла или саркоидоз.
Факторами риска амилоидоза могут являться гиперглобулинемия, нарушения функционирования клеточного иммунитета, наследственная предрасположенность
Затронутые группы населения
По оценкам, ежегодно регистрируется около 4000 новых случаев амилоидоза AL, хотя фактическая заболеваемость может быть несколько выше в результате недостаточного диагноза. Хотя считается, что заболеваемость одинакова у мужчин и женщин, около 60% пациентов, поступивших в центры, являются мужчинами. AL амилоидоз отмечается у людей в возрасте 20 лет, но обычно диагностируется в возрасте 50-65 лет.
Люди с риском развития АА амилоидоза включают людей с хроническими воспалительными заболеваниями, такими как ревматический артрит, псориатический артрит, хронический ювенильный артрит, анкилозирующий спондилит у детей, воспалительные заболевания кишечника.
Люди с хроническими инфекционными заболеваниями, такими как туберкулез, проказа, бронхоэктазия, хронический остеомиелит и хронический пиелонефрит, также находятся в группе риска. Вторичный амилоидоз (АА) встречается у менее чем 5% людей с этими состояниями.
Связанные расстройства
Следующие расстройства могут быть связаны с амилоидозом. Амилоидоз может появляться в сочетании или в результате следующих расстройств:
Множественная миелома, лимфома, лимфома Ходжкина, медуллярная карцинома щитовидной железы, болезнь Уиппла, болезнь Крона, остеомиелит, ревматоидный артрит, анкилозирующий спондилит, синдром Рейтера, псориатический артрит, туберкулез, макроболезность, врожденная гнойно-венозная болезнь (врожденная гиперплазия кишечника, наследственный врожденный ригидроцитогенез)
Диагностика
В частности, в случае амилоидоза AL ранняя диагностика является ключом к выживанию и восстановлению качества жизни после лечения. Диагноз амилоидоз подозревается после подробного анамнеза пациента и клинической оценки, но требует аспирации жировой ткани брюшной полости и/или биопсии вовлеченного органа.
Если заболевание подозревается по клиническим признакам, биопсия пораженного органа даст наибольший результат.
Материал для биопсии исследуется под микроскопом и окрашивается красителем, который будет отдавать зеленым цветом, если смотреть на него в поляризационном микроскопе, если присутствует амилоид. Когда амилоидоз диагностируется при биопсии ткани, важно, чтобы пострадавший был дополнительно исследован, чтобы определить, какие органы поражены.
У людей, находящихся на длительном диализе или с терминальной стадией почечной недостаточности, могут проводиться лабораторные анализы, которые могут анализировать образцы крови или мочи для выявления повышенных уровней белка B2M.
Стандартные методы лечения
В большинстве случаев лечение амилоидоза проводится в домашних условиях. При наличии осложнений больному может быть показана госпитализация.
Терапия амилоидоза включает в себя приём препаратов и соблюдение ряда рекомендаций врача. Но в тяжелых случаях проводится удаление селезёнки, может потребоваться трансплантация почек или печени.
Перечень лекарств зависит от локализации отложений, степени поражения организма, существующих осложнений. Так, при вторичном амилоидозе необходимо специфическое лечение первичного заболевания. Кроме того, назначаются препараты для устранения симптоматики.
Также больному нередко показана специальная диета (ограничение приёма белка и соли).
Специфической профилактической программы амилоидоза не существует, так как точные причины возникновения заболевания неизвестны.
Прогноз
Прогноз зависит от типа амилоидоза и пораженной системы органов, но при соответствующем патогенетическом лечении и поддерживающей терапии продолжительность жизни многих пациентов достаточно велика.
Средняя продолжительность жизни больных с АА-амилоидозом предположительно 10 лет. Наиболее частая причина летального исхода — почечная недостаточность. Нелеченные больные с AL-амилоидозом живут около года от постановки диагноза. Прогноз ухудшает поражение сердечно-сосудистой системы.

Амилоидоз (амилоидная дистрофия) — гетерогенное заболевание, связанное с нарушением белкового обмена, сопровождающееся образованием и отложением в тканях специфического белково-полисахаридного комплекса — амилоида.
Амилоидоз может быть приобретенным или наследственным. Заболевание может быть локализованным или системным. Амилоид может аккумулироваться в печени, почках, сердце, нервах, и кровеносных сосудах, причиняя различные клинические синдромы, включая кардиомиопатию, гепатомегалию, протеинурию, макроглоссию, вегетативную дисфункцию, невропатию, почечную недостаточность, гипертензию.

Амилоидоз, Узел, Конго Красный
- AL-амилоидоз (immunoglobulin light chains derived) — первичный амилоидоз.
- AA-амилоидоз (acquired) — вторичный амилоидоз.
- AF-амилоидоз (средиземноморская перемежающая лихорадка) — наследственная форма амилоидоза, с аутосомно-рецессивным механизмом передачи.
- AH-амилоидоз (hemodialisis-related) — наблюдается исключительно у больных, находящихся на гемодиализном лечении.
- AE-амилоидоз — форма местного амилоидоза, развивающаяся в некоторых опухолях.
- ASC1-амилоидоз — старческий системный амилоидоз.
- Аβ-амилоидоз — при болезни Альцгеймера.
В данной статье более подробно будут рассмотрены два наиболее часто встречающихся вида амилоидоза: первичный и вторичный.
Итак, что же такое первичный амилоидоз или AL-амилоидоз?
Амилоидный амилоидоз легкой цепи (AL), первичный системный амилоидоз (PSA) или просто первичный амилоидоз. Заболевание возникает, когда клетки человека, продуцирующие антитела (плазмоциты), не функционируют должным образом и производят аномальные белковые волокна из компонентов антител, называемых легкими цепями. Эти легкие цепи образуют амилоидные отложения, которые могут нанести серьезный ущерб различным органам. Аномальные легкие цепи в моче иногда называют “Белок Бенс-Джонса”.
AL-амилоидоз является наиболее распространенным типом системного амилоидоза в развитых странах с предполагаемой заболеваемостью 9 случаев на миллион жителей в год. Средний возраст диагностированных пациентов составляет 65 лет и менее 10% пациентов моложе 50 лет.
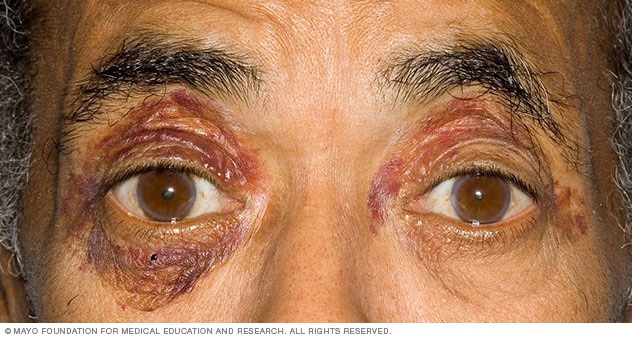
AL-амилоидоз может влиять на широкий диапазон органов, и ,следовательно, проявляется рядом симптомов. Почки являются наиболее часто поражемым органом при АL-амилоидозе. Симптомы заболевания почек и почечной недостаточности могут включать застой жидкости, отек и одышку. Сердечные осложнения, которые затрагивают более трети пациентов, включают сердечную недостаточность и нерегулярное сердцебиение. Другие симптомы могут включать желудочно-кишечные расстройства, увеличение печени, угнетение функции адреналовых и других инкреторных желез, изменение цвета кожи, усталость и потерю веса.

Системный АL-амилоидоз. A.Макроглоссия с боковым гребешком языка. B. Двусторонняя периорбитальная пурпура. C.Псевдо-спортивный вид вторичных диффузных мышечных инфильтратов. D.Объемистая гепатомегалия из-за первичного печеночного амилоидоза. E.Диффузные двусторонние интерстициальные заболевания легких. F.Увеличение подчелюстной железы. Локализованный АL-амилоидоз. G. Узловой конъюнктивный амилоидоз. H.Горатнный амилоидный комок.
Диагноз основывается на исследовании вовлеченного в патологический процесс участка, показывающего Конго красноположительные амилоидные отложения, которые окрашивают положительным антителом анти-LC иммуногистохимией и / или иммунофлуоресценцией. В связи с системным характером заболевания, неинвазивные биопсии, такие как аспирация брюшного жира, должны быть рассмотрены перед взятием биопсий из вовлеченных органов, чтобы уменьшить риск кровотечений.
Наиболее эффективное лечение — аутологичные трансплантации костного мозга с помощью стволовых клеток. Однако не всем пациентам предписывают такой вид лечения.
Другие методы лечения могут включать применение химиотерапии, аналогичной той, которая используется при множественной миеломе. Комбинация мелфалана и дексаметазона была найдена эффективной у тех, кто не подходит для трансплантации стволовых клеток, и комбинация бортезомиба и дексаметазона в настоящее время широко распространена в клиническом использовании.
Продолжительность жизни при АL-амилоидозе зависит от спектра поражения органов (основным фактором прогноза является амилоидная болезнь сердца), тяжести поражения отдельных органов и гематологического ответа на лечение.
Далее рассмотрим вторичный или AA-амилоидоз.
Амилоидоз-АА также известен как вторичный амилоидоз, или реактивный внутрирастительный амилоидоз. Он возникает у пациентов с пролонгированными, хроническими воспалительными или инфекционными заболеваниями на протяжении нескольких лет.
У большинства пациентов с амилоидозом-АА обнаруживаются отложения амилоида в почках, что нарушает структуру почки и вызывает расстройство функции почки, наиболее часто проявляется появлением протеина в моче. В селезенке всегда имеются амилоидные отложения, количество которых может быть увеличено. Могут также быть отложения амилоида в других частях тела, которые обычно не вызывают симптомов.
Фибриллы обнаруженные в амилоидных отложениях образуются из белка, называемого амилоидным А белком, который является производным от нормального кровяного белка неизвестной функции, называемого белком сывороточного амилоида А (SАА). SАА, который вырабатывается в печени, содержится в очень малых количествах (менее 5 мг на литр) в крови всех здоровых людей. Но в ответ на множество видов воспаления, инфекции или ушибы тела, продукция SAA значительно увеличивается, вместе с продукцией другого нормального протеина — C-реактивного протеина (CRP). Производство некоторых других белков крови также увеличивается, хотя и в гораздо меньшей степени, как часть этой нормальной реакции на болезнь, которая называется реакцией острой фазы.
Воспалительные заболевания, которые чаще всего приводят к амилоидозу АА, относятся к следующим категориям.
Ревматологические заболевания, в том числе: ревматоидный артрит, ювенильный артрит, анкилозирующий спондилит и псориатический артрит.
Воспалительные заболевания желудочно-кишечного тракта, включая болезнь Крона и язвенный колит.
Хронические инфекции, такие как: туберкулез, бронхоэктатическая болезнь, остеомиелит, или инфекции, связанные с муковисцидозом, СПИД.
Злокачественные заболевания, включая болезнь Ходжкина, рак почки и болезни Кастельмана.
Наследственные расстройства, которые вызывают нарушение воспалительных генов, как: семейная Среднеземноморская лихорадка (FMF).
Наиболее распространенные симптомы амилоидоза АА вызваны отложениями амилоида в почках. Сначала отложения вызывают нарушение функции почек, а со временем могут привести к конечной стадии почечной недостаточности.
Симптомы болезни почек может включать в себя:
• протеинурия (белок в моче);
• потеря веса и ухудшение аппетита;
• тошнота и рвота;
•повышенная потребность в мочеиспускании на ранних стадиях.
Амилоидоз АА обычно вызывает увеличение селезенки, которое часто не вызывает никаких симптомов, но может быть обнаружено при осмотре врачом. Редко: отложения амилоида АА в щитовидной железе могут вызвать зоб. Отложения амилоида АА в печени могут вызвать увеличение печени. Могут быть отложения амилоида АА в кишечнике и надпочечниках. Эти обычно не вызывает никаких симптомов, но иногда в редких случаях, они могут иметь серьезные последствия. Амилоид АА в сердце очень редко имеет клинически значимые эффекты.
Пациенты с амилоидозом АА обычно обращаются к врачу из-за нарушений функции почек. Более чем в 90% случаев, протеинурия (белок в моче) является первым признаком.
Таким образом, врачи могут заподозрить амилоидоз АА, если у пациента с давним воспалительным состоянием развиваются симптомы заболевания почек, особенно если есть также увеличенная селезенка. Другие признаки заболевания почек при амилоидозе АА могут включать:
— большое количество белка в моче (>3,5 г / сут);
— низкий уровень альбумина в крови;
— периферический отек – опухшие лодыжки.
— гематурия (кровь в моче);
— дефекты почечных канальцев;
— нефрогенный несахарный диабет;
— кальцификация почек (отложения кальция в почках).
Диагноз амилоидоза может быть подтвержден (или может быть устранен) путем взятия биопсии из почки и / или прохождения сканирования SAP. Сканирование SAP показывает распределение и количество амилоида в органах по всему телу. Сканирование SAP произвело революцию в понимании естественного течения амилоидоза и его реакции на лечение.Биопсии могут показать микроскопические следы амилоида в небольших образцах тканей, но не могут обеспечить обзор всего тела. Сканирование SAP является единственным способом получить полную картину степени заболевания. При амилоидозе АА повторное последовательное сканирование SAP может быть очень полезно, показывая, что амилоидные отложения регрессируют (сокращаются), когда эффективно лечится основное воспалительное заболевание.
Лечение всех видов амилоидоза в настоящее время основано на следующих принципах:
1.Сокращение средств обеспечения для формирования амилоидного белка-предшественника(для этого применяют Колхицин).
2.Поддержание функций органов содержащих амилоид.
Подтип АА не лечится.
В 20-е гг. XX столетия Бенхольд предложил окрашивать амилоид конго-красным, затем был обнаружен эффект двойного лучепреломления в поляризованном свете — изменение кирпично-красной окраски на яблочно-зеленую. В 1959 г. Коген и Калкинс с помощью электронной микроскопии установили фибриллярную структуру амилоида.
В нашей стране большой вклад в развитие представлений об амилоидозе внесли Е. М. Тареев, И. Е. Тареева, В. В. Серов. Огромная роль в изучении первичного и генетических вариантов амилоидоза и периодической болезни принадлежит О. М. Виноградовой, чьи монографии, изданные в 1973 и 1980 гг., не утратили своей актуальности и в наши дни.
В настоящее время амилоидоз принято клинически разделять на системные и локальные формы. Среди системных форм, в зависимости от состава фибриллярных отложений, выделяют четыре типа (табл. 1).
К локальным формам амилоидоза в настоящее время относят болезнь Альцгеймера (A-бета, фибриллы состоят из β-протеина, откладывающегося в головном мозге), амилоидоз островков поджелудочной железы, возможно, имеющий патогенетическую связь с диабетом 2 типа, амилоидоз, возникающий в эндокринных опухолях, амилоидные опухоли кожи, назофарингеальной области, мочевого пузыря и другие редкие виды.
Развитие AL-амилоидоза возможно при миеломной болезни, болезни Вальденстрема, В-клеточных лимфомах, и оно может быть идиопатическим при первичном амилоидозе. Все эти варианты объединены общим патогенезом, первичный амилоидоз представляет наибольшую трудность для распознавания в связи с отсутствием явных признаков гематологического заболевания, поэтому именно на данной форме стоит остановиться подробно.
При первичном амилоидозе, доброкачественной плазмоклеточной дискразии, родственной множественной миеломе, аномальные клоны плазматических клеток костного мозга продуцируют амилоидогенные иммуноглобулины. Некоторые аминокислоты в вариабельных участках легких цепей этих иммуноглобулинов занимают необычную позицию, что приводит к их нестабильности и обусловливает склонность к фибриллогенезу. У больных с первичным амилоидозом содержание плазматических клеток в костном мозге повышено до 5—10% (в норме их менее 4%, при миеломной болезни — более 12%), и они продуцируют определенный изотип легких цепей иммуноглобулинов, преобладающий при иммуногистохимическом окрашивании. Свободные моноклональные легкие цепи преобладающего лямбда- или (реже) каппа-изотипа определяются в крови и в моче, но содержание их ниже, чем при миеломной болезни.
Клиническая картина первичного амилоидоза многообразна и определяется преимущественным вовлечением в патологический процесс тех или иных органов — сердца, почек, нервной системы, желудочно-кишечного тракта, печени и др. Первыми симптомами являются слабость и потеря веса, но на этой стадии, до появления органных симптомов, диагноз устанавливается крайне редко.
Органами-мишенями при AL-амилоидозе чаще всего становятся почки и сердце. Поражение почек проявляется нефротическим синдромом, персистирующим и при наступлении ХПН, гематурия и артериальная гипертензия не характерны.
При отложении амилоида в миокарде развиваются разнообразные нарушения ритма, прогрессирующая сердечная недостаточность, чему могут предшествовать бессимптомные изменения на ЭКГ в виде снижения вольтажа зубцов. Эхокардиографическое исследование выявляет концентрическое утолщение стенок левого и правого желудочков, уменьшение объема полостей сердца, умеренное снижение фракции выброса, диастолическую дисфункцию миокарда левого желудочка.
Часто отмечаются симптомы вовлечения нервной системы — вегетативной, в виде ортостатической гипотензии, и периферической — в виде расстройств чувствительности. В последние годы стали описывать также поражения ЦНС, хотя ранее считалось, что они не характерны для первичного амилоидоза.
Диспептические явления (ощущение переполнения, запоры, поносы) и синдром нарушенного всасывания могут быть обусловлены как поражением вегетативной нервной системы, так и амилоидозом желудочно-кишечного тракта. Очень характерна гепатомегалия, природу которой следует дифференцировать между застойными явлениями вследствие сердечной недостаточности и амилоидным поражением печени. Последнее подтверждается повышением уровня щелочной фосфатазы сыворотки крови. Селезенка поражается часто, однако спленомегалия обнаруживается не всегда и большого клинического значения не имеет.
Макроглоссия, классический признак первичного амилоидоза, отмечается у 20% пациентов, инфильтрация мягких тканей может приводить к атрофии мышц, кожи, дистрофии ногтей, алопеции и появлению опухолевидных образований — амилоидом.
Амилоидоз легких часто обнаруживается лишь при аутопсии. Однако в некоторых случаях одышка, кровохарканье и гидроторакс могут быть обусловлены не только застойной сердечной недостаточностью и нефротическим синдромом, но и амилоидным поражением легких. Возможны отложение амилоида в альвеолах и развитие легочных амилоидом. Рентгенологически могут выявляться сетчатые и нодулярные изменения в легочной ткани.
Поражение надпочечников может привести к надпочечниковой недостаточности, нередко остающейся нераспознанной, так как гипотензия и гипонатриемия рассматриваются как симптомы сердечной недостаточности и поражения вегетативной нервной системы. У 10—20% больных может иметь место гипотиреоз как проявление поражения щитовидной железы, нередко встречается увеличение подчелюстных слюнных желез.
Диагноз первичного амилоидоза помимо указанных клинических черт, которые могут быть сходными и при вторичном амилоидозе, базируется на ряде лабораторных данных. У 85% пациентов при иммуноэлектрофорезе белков сыворотки крови и мочи выявляются моноклональные иммуноглобулины. При рутинных исследованиях те же моноклональные иммуноглобулины обнаруживаются в моче в виде белка Бенс-Джонса. Биопсия костного мозга позволяет провести дифференциальный диагноз с множественной миеломой, а также выявить умеренное повышение количества плазматических клеток и их моноклональность при иммуногистохимическом окрашивании.
Однако даже сочетания характерной клинической картины и наличия моноклональных плазмоцитов и белков еще недостаточно для подтверждения диагноза первичного амилоидоза. Решающую роль здесь играют данные биопсии. Наименее инвазивной является аспирация подкожной жировой клетчатки передней брюшной стенки, дающая 80—90% положительных результатов при AL-амилоидозе (в нашей стране этот метод пока не нашел применения). Определенное диагностическое значение имеет биопсия десны и слизистой оболочки прямой кишки, но процент положительных результатов широко варьирует, в зависимости от стадии процесса, поэтому целесообразно выполнение биопсии одного из пораженных орга-нов — почки, печени, сердца, дающее почти 100% положительных результатов при амилоидозе AL-типа.
В первую очередь биопсийный материал окрашивается конго-красным. При обнаружении конгофилии исследуемого материала необходимо его исследование в поляризованном свете, эффект двойного лучепреломления характерен только для амилоида, другие конгофильные вещества яблочно-зеленой окраски не приобретают. После этого желательно типирование амилоида. Наиболее точным является иммуногистохимический метод с использованием моноклональных антител к белкам-предшественникам амилоида. Однако в настоящее время в нашей стране он практически недоступен. Поэтому для диагностики используется окраска с помощью растворов щелочного гуанидина или перманганата калия, что позволяет, хотя и косвенно, определить тип фибриллярных отложений.
Прогноз при первичном амилоидозе хуже, чем при других формах заболевания, средняя продолжительность жизни не превышает двух лет, при наличии поражения сердца или мультисистемного поражения без лечения больные погибают в течение нескольких месяцев. Наиболее частыми причинами смерти являются сердечная и почечная недостаточность, сепсис, сосудистые осложнения и кахексия. Патогенетическое сходство с миеломной болезнью позволяет рассчитывать на торможение прогрессирования заболевания при химиотерапии, проводимой с целью подавления моноклональных плазмоцитов. Существует несколько схем лечения (табл. 2).
Применение химиотерапии в случае успеха лечения позволяет увеличить продолжительность жизни больных на срок от 10 до 18 мес. Но эффективность терапии невысока, в частности, в связи с тем, что во многих случаях прогрессирование заболевания приводит к гибели больных до завершения курса лечения, а также из-за развития цитопении, инфекционных осложнений, фатальных нарушений ритма при лечении сверхвысокими дозами дексазона. Применение высоких доз мельфолана с трансплантацией аутологичных стволовых клеток позволяет достичь ремиссии более чем в 50% случаев, однако использование этого метода ограничено тяжестью состояния, возрастом больных, функциональными нарушениями со стороны сердца и почек. Во многих случаях возможна лишь симптоматическая поддерживающая терапия.
Развитие AA-амилоидоза происходит при хронических воспалительных процессах, предшественниками AA-амилоида являются сывороточные острофазовые белки, α-глобулины, продуцируемые клетками разных типов, в основном нейтрофилами и фибробластами. Вторичный амилоидоз развивается при ревматоидном артрите, болезни Бехтерева, псориатическом артрите, различных опухолях, лимфогранулематозе, неспецифическом язвенном колите и болезни Крона, при периодической болезни (семейной средиземноморской лихорадке), а также при туберкулезе, остеомиелите, бронхоэктатической болезни.
Характерными клиническими особенностями АА-амилоидоза является поражение почек у большинства пациентов, а также относительно редкое поражение печени и/или селезенки (около 10%) и сердца (выявляется лишь при эхокардиографии). Макроглоссия для вторичного амилоидоза не характерна. Диагноз основан на сочетании амилоидоза почек и хронического воспалительного заболевания, подтверждением служит иммуногистохимическое окрашивание биопсийного материала, в нашей стране используются уже упомянутые выше косвенные окрасочные методы.
Прогноз во многом зависит от природы основного заболевания, при естественном течении у трети больных через 5 лет от момента выявления протеинурии развивается почечная недостаточность. При периодической болезни пятилетняя выживаемость составляет 25%.
Лечение основано на подавлении очага — источника продукции сывороточных белков-предшественников. Удаление опухолей, секвестрэктомия, резекция кишки, лечение туберкулеза, уменьшение активности ревматоидного артрита (при использовании цитостатиков) приводят к прекращению прогрессирования амилоидоза, а иногда и к обратному развитию клинических проявлений, в частности нефротического синдрома.
Применение колхицина при периодической болезни является методом выбора, эффективность его доказана, лечение предотвращает развитие амилоидоза и тормозит его прогрессирование. При других формах вторичного амилоидоза эффективность колхицина не подтверждена.
Сенильные и наследственные формы системного амилоидоза, так же как и локальные формы, встречаются редко, диализный амилоидоз хорошо известен специалистам, в общей практике с ним сталкиваться практически не приходится.
Симптоматическая терапия зависит не от типа амилоидоза, а от пораженных органов-мишеней (табл. 3).
Амилоидоз, особенно первичный, считается нечастой патологией, однако в действительности он не столько редко встречается, сколько с трудом диагностируется. Адекватная диагностика требует не только знания клиники и патогенеза данного заболевания, но и наличия определенных диагностических возможностей. Чтобы проиллюстрировать это положение, приведем собственные данные (см. таблицу 4). В нефрологическом отделении МГКБ имени С. П. Боткина в 1993—2003 гг. наблюдалось 88 больных, которым был поставлен диагноз амилоидоза.
Диагноз был подтвержден морфологически у всех больных с AL-амилоидозом, старческим и неуточненным по типу амилоидозом, и у 30 пациентов со вторичным амилоидозом — всего в 53 случаях. У 12 больных выполнялась биопсия почки, а у двоих — биопсия печени, у восьми — биопсия кишки, в 12 случаях — десны, еще в 19 случаях диагноз был подтвержден при морфологическом исследовании секционного материала.
В большинстве случаев диагноз амилоидоза был установлен впервые в результате обследования в нефрологическом отделении. Нами было проведено сопоставление среди больных с AL-амилоидозом направительного и клинического диагнозов (табл. 5).
Все больные, у которых диагностировалась миеломная болезнь с развитием AL-амилоидоза, были переведены в гематологические отделения. Из 11 больных с первичным амилоидозом семь пациентов получали химиотерапию комбинацией мельфолана с преднизолоном внутрь прерывистыми курсами, четверо из них — в сочетании с диализным лечением, и еще одна больная — только диализное и симптоматическое лечение. Из числа этих больных пять человек умерли в сроки от двух недель до двух лет от начала лечения (все с почечной недостаточностью и полиорганным поражением), один больной находится на диализе, один больной был направлен на трансплантацию аутологичных стволовых клеток, и одна больная получает лечение до настоящего времени. У одного пациента химиотерапия отложена в связи с наличием длительно не рубцующейся язвы желудка, и еще двое больных отказались от лечения.
Среди больных с вторичным амилоидозом в нашем исследовании преобладали пациенты с ревматоидным артритом, на втором месте среди причин — хронический остеомиелит и псориатический артрит, остальные заболевания встречались реже (табл. 6).
Лечение ревматоидного артрита и псориатического артрита проводилось с применением цитостатиков (метатрексата, азатиоприна), хотя во многих случаях возможности терапии были ограничены из-за наличия ХПН и сопутствующей патологии. Больные с хроническим остеомиелитом были направлены в отделения гнойной хирургии. Пациенты с болезнью Бехтерева и болезнью Крона получали специфическое лечение, больные с ХНЗЛ и туберкулезом также были направлены в профильные стационары. Одна из больных с опухолью желудка была успешно оперирована, и на протяжение четырех лет наблюдения нефротический синдром постепенно регрессировал, в остальных случаях опухолей распространенность процесса позволяла проводить только симптоматическую терапию, больной с лимфогранулематозом поступил в терминальном состоянии. Смертность среди пациентов со вторичным амилоидозом составила 38% (за счет больных с далеко зашедшим поражением на момент постановки диагноза). Все больные с периодической болезнью получали терапию колхицином.
При осмотре — кожа чистая, обычной окраски, анасарка, отеки массивные, плотные, определяется асцит, периферические лимфатические узлы не увеличены. АД 110/70 мм рт. ст., тоны сердца звучные, ясные, ритмичные, ЧСС 90 уд/мин, печень и селезенка не увеличены, диурез до 1000 мл/сут, стул регулярный, без патологических примесей. При обследовании выявлен нефротический синдром — протеинурия 3 г/л, мочевой осадок скудный, гиподиспротеинемия, гиперлипидемия (общий белок сыворотки крови 39 г/л, альбумины 12 г/л, глобулины 7-30-15-19% соответственно α1-α2-β-γ холестерин 17,8 ммоль/л, β-липопротеиды 250 ЕД), при анализе мочи на белок Бенс-Джонса — реакция отрицательная, суточная экскреция 17-КС не снижена. Клинический анализ крови и другие биохимические показатели в пределах нормы, коагулограмма — выраженная гиперфибриногенемия, повышение уровня РКФМ. Исследование иммуноглобулинов крови: Ig-A — 0,35, Ig-M — 35,7 (две нормы), Ig-G — 1,96 г/л. Рентгенография органов грудной клетки, костей черепа и таза, УЗИ брюшной полости, почек, щитовидной железы, ЭХО-КГ без патологии, УЗИ малого таза — признаки аденомиоза тела матки, ЭГДС — рефлюкс-эзофагит, хронический гастрит. При осмотре невропатологом патологии не найдено, онкологом установлена фиброзно-кистозная мастопатия.
С целью уточнения генеза нефротического синдрома под местной анестезией под УЗ-наведением выполнена тонкоигольная пункционная биопсия правой почки, осложнений не было. При исследовании биоптата в мезангии клубочков и во внегломерулярных сосудах отмечается отложение амилоида. Амилоид загружает до 25% сосудистых петель клубочков. При иммуногистохимическом исследовании специфической люминисценции не найдено. При обработке препаратов раствором щелочного гуанидина в течение 2 ч конгофилия и их свойства в поляризованном свете сохраняются, что характерно для AL-амилоидоза.
Установлен диагноз первичного амилоидоза с поражением почек, нефротическим синдромом, сохранной почечной функцией, признаков иных органных поражений не выявлено. С января 2003 г. начата химиотерапия мельфоланом 16 мг/сут и преднизолоном 100 мг/сут, курсами по четыре дня каждые шесть недель. Проводится также симптоматическое лечение: фуросемид, верошпирон, препараты калия, фамотидин, переливания альбумина. К настоящему времени проведено пять курсов химиотерапии с хорошей переносимостью, отеки уменьшились, протеинурия снизилась до 1,8 г/л, несколько уменьшилась выраженность гиподиспротеинемии (общий белок 46 г/л, альбумины 18 г/л, α2-глобулины 20%). Функция почек остается сохранной, креатинин плазмы 1,3 мг/Дл, признаков поражения других органов и систем при контрольных динамических обследованиях не выявлено.
В заключение отметим, что амилоидоз представляет собой тяжелое заболевание с высоким уровнем летальности, которое чрезвычайно трудно диагностировать, однако своевременное и качественное обследование больных позволяет поставить диагноз в более ранние сроки, а своевременное назначение адекватной терапии, в свою очередь, дает возможность улучшить прогноз в этой группе больных.
Е. В. Захарова
Московская городская клиническая больница им. С. П. Боткина
Читайте также:
