
Атаксии телеангиэктазии вследствие дефекта т-клеток что это
Атаксия телеангиэктазия — это редко встречающаяся наследственная патология, которая дополняется частым поражение организма респираторными инфекциями и склонностью к развитию опухолей злокачественного характера.
Почему и каким образом развивается болезнь
Основой болезненных изменений, которые дополняют заболевание, являются нарушения генетики, что провоцирует формирование врожденной дисплазии. Сидром Луи-Бар — это заболевание, протекающее по аутосомно-рецессивному типу, то есть проявляется себя лишь при условии проникновения рецессивного гена одновременно от обоих родителей.
Это важно! Морфологически для данной патологии характерны дегенеративные изменения в тканях мозжечка, обычно это утрата им особых клеток. Порой дегенеративные нарушения влияют на мозжечковое зубчатое ядро, на черную субстанцию и некоторые части коры головного мозга.
Синдром дополняет гипоплазию или аплазию тимуса и врожденным недостатком некоторых элементов. Такие отклонения в работе иммунитета вызывают частые заражения организма инфекциями, которые долго не лечатся и вызывают множество осложнений. Нарушения иммунитета способны стать причиной формирования злокачественных опухолей, начинающихся в лимфоретикулярной системе.
Как патология проявляет себя в человеческом организме
Чаще всего синдром клинически начинается давать о себе знать в возрасте до трех лет. В любой ситуации патология прогрессирует одновременно с появлением мозжечковой атаксии. Её симптомы будут очевидны лишь в момент, когда ребенок научится ходить. Таким образом, становятся заметны нарушения походки и координации движений, дрожи в процессе двигательного акта, качание головой и качание туловищем.
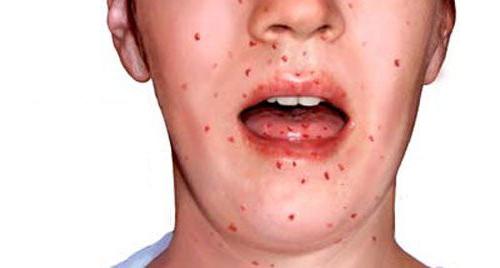
Проявление телеангиэктазии или попросту сосудистых звездочек, которые сопровождают данное заболевание, начинается уже с возраста ребенка от трех — шести лет. Обычно это небольшие пятна красноватого или розоватого оттенка разной формы, имеющие несильные разветвления, наличие которых объясняется расширением маленьких сосудов кожного покрова.
Для синдрома Луи-Бар характерно первоначальное формирование сосудистых звездочек на конъюнктиве глазного яблока в виде так называемых паучков. Впоследствии звездочки охватывают кожный покров носа, век, шеи и лица, сгибов локтей и колен, тыльную поверхность кистей и стоп.
Помимо сосудистых звездочек кожные симптомы включают в себя формирование пигментных пятен светло-коричневого оттенка, формирование веснушек и областей обесцвеченной кожи.
Это важно! Также для синдрома Луи-Бар становится характерно частое поражение инфекциями путей дыхания, потому что ухудшение работы иммунитета в основном вызывает часто возникающие инфекции путей дыхания и уха — фарингит, хронический ренит, бронхит, воспаление легких, синусит, отит.
Среди людей, страдающих этой болезнью, процессы развития злокачественных опухолей диагностируются во много раз чаще по сравнению со средними показателями здорового населения.
Проведение лечения патологии
К сожалению, современная медицина ещё не в состоянии разработать эффективные методы терапии для синдрома Луи-Бара, поэтому методы до сих пор остаются предметом поиска специалистов.
В современной медицине могут использоваться только способы паллиативной симптоматической терапии для нарушений работы иммунитета и соматических нарушений.
Жизнь пациентов с синдромом Луи-Бара продляется благодаря корректирующей иммунитет терапии посредством тимуса, витаминных комплексов в больших дозах. Также организуется интенсивное лечение любого инфекционного процесса. По показаниям врача назначаются противовирусные средства, антибиотические препараты широкого спектра действия, противогрибковые препараты и глюкокортикостероиды.
Не разрешается самостоятельно принимать препараты или давать препараты ребенку без назначения врача. Самолечение может привести к серьезным последствиям и ухудшению состояния здоровья.
Атаксия телеангиэктазия, известная также как синдром Луи-Бара является редким нейродегенеративным наследственным заболеванием, вызывающим тяжёлую инвалидность. Атаксия приводит к плохой координации, а телеангиэктазия — слегка расширенным кровеносным сосудам; оба признака являются отличительными чертами болезни. Атаксия телеангиэктазия вызывается мутациями в гене ATM, который отвечает за ответ клетки на двунитевые разрывы в ДНК.
При атаксии телеангиэктазии страдают различные органы и функции организма. Во-первых, при этом заболевании ухудшается работа определённых областей мозга, включая мозжечок, что вызывает трудности с движениями и координацией. Во-вторых, при АТ ослаблена иммунная система, что приводит к предрасположенности к инфекции. В-третьих, при АТ нарушена репарация ДНК, что увеличивает риск развития опухолей.
Описание синдрома
Синдром Луи-Бар впервые был описан в 1941 году во Франции. Нет точных данных о том, с какой частотой синдром Луи-Бар встречается среди современного населения. По некоторым сведениям эта цифра составляет 1 случай на 40 тысяч новорожденных. Однако, необходимо учитывать, что при смерти в раннем детском возрасте синдром Луи-Бар обычно остается не диагностированным. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологиисиндром Луи-Бар относится к так называемым факомотозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.

Причины развития патологии
Данная аномалия развития закладывается еще в первом триместре беременности. Заболевание передается только по наследству. Синдром относится к аутосомно-рецессивным генетическим патологиям. Это означает, что ребенок точно унаследует заболевание, если оба родителя имеют нарушение хромосом. Если же аномалия наблюдается у одного из них (независимо от пола), то шанс появления синдрома Луи-Бар у малыша составляет 50%. Основная причина мутации – это нарушение длинного плеча 11-й хромосомы. Точные факторы, которые приводят к такой генетической перестройке, неизвестны. Но выделяют ряд вредных воздействий, влияющих на эмбриональное развитие. В первую очередь это факторы окружающей среды (облучение, отравление ядовитыми веществами). Также в первом триместре беременности очень опасен стресс.
Клинические проявления
Это редкое заболевание. Первая симптоматика проявляется в возрасте от трех месяцев до трех лет. С возрастом проявления становятся более выраженными.
Телеангиэктазия дебютирует в основном после признаков атаксии в возрасте 4-6 лет. Бывают случаи, когда симптоматика наблюдается уже на первом месяце жизни. Телеангиэктазии проявляют себя в первую очередь на глазных яблоках в виде бульбарной коньюктивы, далее распространяется на веки и лицо.
Характерные симптомы синдрома Луи-Бар:
- Нарушения координации движений (обычно после трех лет) — неустойчивость, атаксическая походка, непроизвольные движения;
- Нарушения психики и замедление или полная остановка в развитии (после десяти лет);
- Изменение цвета кожи под воздействием ультрафиолетовых лучей;
- Образование былых пятен на теле;
- Расширение кровеносных сосудов в области внутренней стороны коленей и локтей, на лице, в белках глаз;
- Ранняя седина;
- Повышенная чувствительность к рентгеновским лучам;
- Тяжелые инфекции дыхательных путей, ушей, склонные к рецидиву (у 80% больных);
- Отсутствие рефлексов в мышцах глаз;
- Аномальное развитие вилочковой железы, а в некоторых случаях и ее полное отсутствие;
- Лимфоцитопения (примерно 1/3 всех случаев);
- Задержка полового развития или неполное развитие и ранняя менопауза.
Дерматологические проявления у больных синдромом Луи-Бар наблюдаются в 100% случаев. Иные проявления типа сухости кожи, кератоза на кожных покровах конечностей, пигментация на лице встречаются примерно в половине случаев. Нельзя сказать, что кожные проявления специфичны для атаксии-телеангиэктазии, но это первый видимый признак заболевания, что является очень важным для своевременной и правильной диагностики и лечения. Часто именно дерматологическая картина помогает установить правильный диагноз.

Диагностика
Постановка диагноза атаксии-телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться:
- УЗИ тимуса,
- МРТ головного мозга,
- фарингоскопия,
- риноскопия,
- рентгенография легких.
При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений.
Лечение
Для лечения этого редкого заболевания необходимо соблюдать несколько принципов лечения, которое должно быть комплексным и этиологическим, патогенетическим, индивидуальным:
- Антибиотикотерапия позволит бороться с вторичной инфекцией бактериального происхождения, которая развивается при выраженном иммунодефиците.
- Для повышения защитных свойств организма специалист назначает иммуностимуляторы, гамма-глобулины, витаминотерапию и общеукрепляющие лекарственные средства.
- Физиотерапевтические мероприятия.
- Посещение логопеда для постановки речи ребенку.
- При наличии опухолей проводится лучевая терапия, химиотерапия или хирургическое удаление новообразования.
- Если основным заболеванием является сахарный диабет, то обязательно назначение инсулина или противодиабетических лекарственных средств.
Прогноз
В связи с отсутствием эффективного лечения максимальный срок жизни пациентов, с диагнозом синдром Луи Бара не превышает 20 лет. Однако, даже до такого возраста доживают лишь единицы. Злокачественные новообразования, серьезные инфекционные заболевания убивают больных намного раньше.
Атаксия телеангиэктазия — это заболевание, при котором отмечается нарушением иммунитета, связанного с Т-клетками в совокупности с мозжечковой атаксией прогрессирующего характера, наличием распространенных телеангиэктазий кожных покровов и глазной конъюктивы. Что является причиной заболевания?
В основе данной редкой патологии лежит генетическая наследственность по аутосомно-рецессивному типу. Второе ее название — синдром Луи-Бар.
Особенностью атаксии телеангиэктазии являются дегенеративные врожденные изменения тканей мозжечка (зернистых клеток и волокон Пуркинье, черной субстанции) и тимуса. Реже отмечаются поражения отдельных участков коры и задние столбы (нервные волокна) спинного мозга. Это и определяет ее основные симптомы — нарастающая дискоординация движения и выраженное снижение иммунных сил организма, которые лежат в основе осложнений данного заболевания — образование опухолей и частые серьезные инфекции.
Как проявляется атаксия телеангиэктазия
Чаще всего синдром проявляется у ребенка от пяти месяцев и до достижения им трехлетнего возраста. Расстройство координации у грудничка диагностировать трудно, но оно становится заметным после того, как малыш пытается начать ходить. У него наблюдается дрожание головы, рук, иногда всего тела. Характерно также изменения в походке, возникновение косоглазия и нистагм.
В редких случаях такие симптомы не проявляются до 4 лет. К 10 годам зачастую передвижение становится невозможным, однако иногда имеются отдельные случаи улучшения состояния вследствие включения механизмов компенсации других структур, отвечающих за двигательную систему.
Также часто при этом синдроме могут быть расстройства речи, снижение мышечного тонуса и гипорефлексия.
Телеангиэктазии заметными становятся от трех до шести лет. Возникают эти сосудистые звездочки из-за нарушения структуры подкожных мелких сосудов. Вначале они локализуются только на бульбарной конъюктиве, а затем переходят на нос и другие части лица и шеи, типично их расположение при такой форме заболевания на тыльной стороне кистей и стоп. 
Иногда кожные проявления атаксии телеангиэктазии настолько выражены, что полностью сливаются на лице, а в сочетании с уплотнением кожи их могут принять за системную склеродермию.
Появление сосудистых звездочек встречается как признак и других заболеваний, но в сочетании с нарушением равновесия и координации они являются специфическим признаком атаксии телеагиэктазии.
Дети с синдромом Луи-Бар часто страдают инфекционными заболеваниями — воспалением легких, бронхитом, отитом, гайморитом. Это объясняется неспособностью организма справляться с микробной и вирусной инфекцией в силу ослабленного иммунитета.
Нередко при такой патологии могут возникать значительные нарушения в работе эндокринных органов. Малыши страдают сахарным диабетом, у мальчиков наблюдается недоразвитие яичек, бесплодие.
В крови отмечается резкое снижение иммуноглобулинов и Т-лимфоцитов. У 40% больных отмечаются аутоиммунные реакции, что выявляется при нахождении в крови специфических антител.
При таком синдроме частота возникновения злокачественных новообразований в 1000 раз выше, чем среди остального населения.
Онкопатология при атаксии теолеангиэктазии часто представлена лейкозами, раком желудка, новообразованиями головного мозга.
Прогноз при атаксии телеангиэктазии неблагоприятный. Все процессы и проявления прогрессируют, развивается выраженная мышечная слабость, паралич, слабоумие. Максимальный возраст жизни таких больных при благоприятных условиях — около 30 лет.
Методы лечения
К сожалению, современный уровень науки не может полностью излечить этот синдром. Поэтому все усилия врачей направляются на коррекцию симптомов и осложнений.
Инфекционные процессы лечатся при помощи антибактериальных средств. Применяются также общеукрепляющие препараты. Есть предложенные научные разработки по хирургической пересадке тимуса, но пока они в практической медицине не используются.
В ряде случаев практикуется трансплантация костного мозга, иммуноглобулинотерапия, введение гормонов вилочковой железы.
Внешние канцерогенные (ионизирующее излучение, некоторые химические вещества) и генетические факторы, предрасполагающие к повышению частоты хромосомных мутаций, одновременно предрасполагают и к возникновению новообразований. Это соответствует общепризнанной концепции клональной природы злокачественных опухолей и подтверждается высокой частотой онкологической патологии у больных с генетическими синдромами, в том числе кожными. Хотя каждый из приведенных здесь генодерматозов носит врожденный характер или проявляется в раннем детстве, многие из них не сопровождаются развитием злокачественных новообразований до наступления взрослого возраста.
Атаксия-телеангиэктазия (син.: синдром Луи-Бар) — редкое системное заболевание, характеризующееся сочетанием хромосомной нестабильности, мозжечковой атаксии, кожно-глазных телеангиэктазий, комбинированным иммунодефицитом и повышенным риском развития опухолей. Тип наследования аутосомно-рецессивный. Мугантный ген локализуется в 11-й хромосоме на участке q22-q23. У гетерозиготных носителей иногда отмечаются дефицит IgE и телеангиэктазий на коже и слизистых оболочках.
Атаксия-телеангиэктазия начинается в раннем детстве и проявляется мозжечковой атаксией (в 100% случаев), дрожанием головы и туловища, нарушением походки, интенционным тремором и хореоатетозом (в 90-100% случаев). Характерны нарушения движения глазных яблок (у 80-90% больных), нистагм (у 90-100%) и косоглазие, а также гипотония мышц, гипорефлексия, дизартрия. Дети отстают в умственном развитии.
В возрасте от 2 до 6 лет появляются телеангиэктазии на конъюнктиве, сгибательных поверхностях конечностей, слизистых оболочках мягкого и твердого нёба, склеродермия, прогерические изменения кожи и волос. Гистологические изменения в коже неспецифичны, отмечаются повышенное содержание пигмента в эпидермисе, расширение капилляров с периваскулярной инфильтрацией из нейтрофилов и лимфоцитов. Также описаны гирсутизм и acantosis nigricans.
Атаксия-телеангиэктазия сопровождается нарушением Т-и В-систем иммунитета, отсутствием сывороточных IgA, а иногда также IgG и IgE. Этим обусловлены хронические респираторные инфекции — синуситы и пневмонии (в 60-80% случаев).
Повышенная частота лейкозов и лимфом связана с хромосомной нестабильностью и другими нарушениями, например, повышенной чувствительностью к ионизирующей радиации. Недавние наблюдения указывают на 3-5-кратное повышение риска развития рака для гетерозиготных носителей гена атаксии-телеангиэктазии и на особенный риск развития рака молочной железы у женщин-носителей.
Диагноз атаксии-телеангиэктазии устанавливается на основании трех основных признаков: мозжечковой атаксии, телеангиэктазий и комбинированного иммунодефицита.
Дифференциальный диагноз атаксии-телеангиэктазии проводится с пигментной ксеродермой. пойкилодермией Ротмунда—Томсона, синдромом Блума, синдромом Коккейна, врожденным дискератозом.
Течение атаксии-телеангиэктазии прогрессирующее. Прогноз неблагоприятный вследствие тяжело протекающих легочных инфекций и повышенного риска злокачественных опухолей.
Лечение атаксии-телеангиэктазии в применении неспецифической иммунотерапии и общеукрепляющих средств, при необходимости — антибиотикотерапии. Рентгенотерапия новообразований противопоказана.

Гирсутизм нижних конечностей и экхимозы, развившиеся вторично вследствие атаксии и падений
Синдром Вискотта—Олдрича
Синдром Вискотта—Олдрича — наследственный симптомокомплекс, характеризующийся тромбоцитопенической пурпурой, экземой и рецидивирующими инфекциями. Поражает мальчиков. Тип наследования рецессивный, связанный с Х-хромосомой. Установлено, что лимфоциты, нейтрофилы и тромбоциты больного изменяются под влиянием сильно гликозированной поверхности протеина сиалоформина (CD43), который не откладывается на других типах клеток. По-видимому, патологическое влияние измененного CD43 на клетки крови обусловлено повышенной чувствительностью этих клеток к протеолитическому расщеплению. Тромбоциты больных способны к нормальной агрегации, но их продукция и время существования сокращены, в результате чего развивается тромбоцитопения. В крови повышен уровень IgE и понижен уровень IgM. Содержание IgA и IgG в пределах нормы или повышено. Лимфоциты также аномальны, их чувствительность к антигенам in vitro снижена при сохранении чувствительности к митогенам. Предполагали, что первичным дефектом синдрома является дефицит синтеза CD43, однако выяснилось, что ген, контролирующий синтез этого протеина, находится в хромосоме 16, а не в Х-хромосоме, и, следовательно, его синтез не нарушен.
Патологический клинический комплекс синдрома вискотта - олдрича проявляется на первом году жизни классической триадой — иммунодефицитом, тромбоцитопенией и экземой.
Экзема при синдроме вискотта - олдрича развивается уже на 1 -й неделе жизни, локализуется на волосистой части головы, лице, в аногенитальной области, на сгибательных поверхностях конечностей и практически не отличается от атопической экземы, за исключением сочетания с пурпурой и возникновением интенсивных кровотечений из экскориаций. Кровотечения наиболее выражены на 6-м месяце жизни, возможны кишечные кровотечения, внутричерепные кровоизлияния. Дети склонны к бактериальным инфекциям (с развитием отита, пиодермии, менингита, септицемии), вирусным заболеваниям. Высока склонность к аутоиммунным процессам с развитием васкулитов, поражений почек (хронический гломерулонефрит), гемолитической анемии, артритов. Харакерны энтеропатии с большой потерей белка, бронхиальная астма, контактные уртикарные сыпи.
Большинство больных синдромом вискотта - олдрича умирают от инфекций или кровотечений в детстве, но у 5-12% в будущем развиваются злокачественные лимфопролиферативные заболевания, наиболее часты болезнь Ходжкина или острый лейкоз.
Лечение синдрома вискотта - олдрича: остановка кровотечений, переливание тромбоцитной массы, спленэктомия, антибактериальная терапия (у -глобулин, антибиотики и др.), трансплантация костного мозга.

- 7 Июля, 2018
- Неврология
- Гладких Ася
Синдром Луи-Бар относится к редким иммунодефицитным нейродегенеративным генетическим заболеваниям, проявляющимся в виде мозжечковой атаксии, которая вызывает паралич тяжелой формы. Для атаксии характерно нарушение координации движений, а для телеангиэктазии — расширение кровеносных сосудов. Два этих признака – это отличительные черты синдрома. Поэтому у заболевания имеется второе название — атаксия-телеангиэктазия.
Тип наследования
Передается данный недуг по аутосомно-рецессивному типу. Риск заболевания ребенка, рожденного в семье, в которой один больной родитель, составляет 50/50. Согласно статистическим данным, заболевание мало распространено, из сорока тысяч оно приходится на одного человека.

Данная патология характеризуется врожденным неправильным иммунным состоянием человеческого организма. Происходит поражение Т-звена в генетической цепочке. Затем болезнь проявляется в виде аномальных форм по всему организму. В связи с поражением иммунитета люди, которые больны синдромом Луи-Бар, подвержены частым инфекционным заболеваниям. Кроме этого, могут возникнуть злокачественные онкологические опухоли по всему телу.
В случаях, когда синдром проявляется у новорожденного ребенка, шансы на выживание практически отсутствуют. Причем во многих случаях не удается даже своевременно и правильно поставить диагноз.
Причины патологии (мнение специалистов)
Синдром Луи-Бар в различных классификациях рассматривают как спинно-мозжечковую дегенерацию или как факоматоз (данный термин предложили в качестве обозначения заболеваний с совмещенным поражением нервной системы и покровов кожи — врожденных нейро-эктомезодермальных дисплазий). Причиной является мутация АТМ гена, активирующего аутоиммунные процессы, которые приводят к уничтожению клеток всего организма, в том числе и головного мозга. Генетические отклонения проявляются уже во внутриутробном периоде.
Данная болезнь в равной степени выявляется и у мужчин, и у женщин. Она стремительно развивается, поражая в первую очередь ЦНС и покровы кожи. Недуг имеет способность полностью изменить или разрушить ткани мозжечка, но поражает его ядро.
Синдром Луи-Бар – это иммунодефицитное состояние, основанное на гипоплазии тимуса и дефиците IgA и IgE. Это означает, что нарушается иммунная деятельность клеточного и гуморального звеньев. Это может провоцировать часто повторяющиеся заболевания дыхательных путей, носящие инфекционный характер, ЖКТ и кожных покровов. Типичная гипоплазия вилочковой железы может быть дополнена гипо/атрофией лимфатических узлов и в целом лимфатического аппарата, а также селезенки и пищеварительного канала.
Ослабленному иммунитету не по силам выстоять даже перед несерьезной инфекцией, кроме того, он становится чувствительным перед злокачественными новообразованиями в лимфатической системе.
Клиника синдрома
Это врожденное заболевание встречается довольно редко. Первые симптомы могут проявиться в возрасте 3-х месяцев и далее - до 3-х лет. По мере роста больного проявления становятся более выраженными.
Телеангиэктазия проявляется, как правило, после признаков атаксии, при достижении больным возраста 4-6 лет. Иногда случается так, что симптомы заметны уже в первые недели жизни. Телеангиэктазии характеризуются проявлениями изначально на глазных яблоках по типу бульбарной коньюктивы, после они начинают распространяться на веки и лицо.
Характерная симптоматика синдрома Луи-Бар:
- нарушается координация движений (после 3-лет);
- страдает психика и замедляется или полностью останавливается развитие;
- изменяется цвет кожи под ультрафиолетовыми лучами;
- образуются белые пятна на теле;
- расширяются кровеносные сосуды в области внутренней стороны коленей и локтей;
- появляется ранняя седина;
- наблюдается повышение чувствительности к рентгеновским лучам;
- прогрессируют тяжелые инфекции дыхательных путей, ушей (у 80% больных);
- отсутствуют рефлексы в мышцах глаз;
- неправильно развивается вилочковая железа, иногда она вовсе отсутствует;
- наличие лимфоцитопении у 1/3 больных;
- половое развитие задерживается, наступает ранняя менопауза.
Это характерно для неврологических болезней.

Проявления симптомов на кожных покровах наблюдаются при данном заболевании в 100% случаев. Возникновение признаков другого типа – сухость кожи, кератоз кожных покровов конечностей, пигментация на лице – можно встретить лишь в половине случаев. Кожные проявления не являются специфичным симптомом для патологии, но это первый наглядный признак болезни, который очень важен при своевременном и правильном диагностировании и лечении. Зачастую именно благодаря ему можно своевременно и правильно поставить диагноз.
Диагностирование синдрома
По мнению специалистов, синдром Луи-Бар распознать может быть сложно по причине сочетания в нем признаков других генетических заболеваний, которые скрывают настоящие симптомы. В большинстве случаев выявить и провести диагностику недуга возможно только по истечении продолжительного лечения инфекционных заболеваний, не дающего результатов.
Чтобы не ошибиться и установить правильный диагноз, больной должен проконсультироваться не у одного медицинского специалиста. Это может быть иммунолог, дерматолог, офтальмолог, онколог, отоларинголог. Нужно проанализировать пройденные процедуры, изучить анализы, прочитать заключения специалистов после консультаций. И только после этого окончательный диагноз сможет поставить врач-невролог. Кроме того, он должен назначить лабораторные исследования, другие процедуры и тесты, это поможет не ошибиться в формулировках.

Что выявляют на осмотре?
При осмотре врач должен обратить особое снимание на:
- замедление темпов развития;
- пигментацию кожных покровов;
- нарушение или отсутствие сухожильных рефлексов;
- нарушение роста;
- уменьшенные размеры миндалин, лимфатических узлов.
На что направлена диагностика?
Список лабораторных анализов, которые могут быть назначены, нацелены на:
- установление уровня белка α-фетопротеина (при заболевании его содержание повышено);
- снижение уровня лейкоцитов;
- исследование уровня иммуноглобулина в крови (при болезни уровни иммуноглобулина А и Е сильно снижены);
- выявление генетических мутаций;
![]()
- определение переносимости глюкозы;
- ультразвуковое обследование вилочковой железы.

Кроме этого, проводится МРТ головного мозга и мозговых структур (при синдроме заметно увеличен четвертый желудочек и наблюдаются изменения патологического уровня в мозжечке — дегенерация мозжечковых клеток). Рентгенография грудной клетки поможет исключить пневмонию.
Терапия заболевания
На сегодняшний день медицина не в состоянии бороться с таким тяжелым генетическим врожденным заболеванием, как синдром Луи-Бар. Решение данного вопроса является одной из главных задач экспериментальной медицины в области генетики. Как правило, лечение заключается в замедлении прогрессирования заболевания и уменьшению его симптомов.
Терапия может быть назначена только врачом-неврологом, причем каждый отдельный случай рассматривается индивидуально для каждого пациента. При этом учитываются этиология, патогенез, стадия заболевания. Чтобы продлить жизнь пациента, специалист назначает особую иммунотерапию с разными дозами Т-активина и гамма-глобулина. Вместе с тем необходимо обязательно принимать витамины, чтобы поддерживать правильное функционирование организма.
Курс антибиотиков
Пациент проходит назначенный ему курс терапии антибиотиками, чтобы побороть вторичную инфекцию, носящую бактериальный характер. Также больному прописываются физиотерапевтические процедуры.
Химиотерапия
Если врач при диагностировании обнаруживает злокачественные новообразования, он назначает химиотерапию, лучевую терапию или ставит вопрос о хирургическом вмешательстве. Если больной страдает сахарным диабетом, то в таком случае ему назначают инсулин и противодиабетические препараты. К неврологическим болезням нужно подходить со всей серьезностью.
Как спрогнозировать синдром?
Так как заболевание является генетическим, разрушает частично или полностью на клеточном уровне иммунитет, носит патологический характер и не излечивается, то естественная полноценная жизнедеятельность становится практически невозможной.

Прогнозы при этом заболевании врожденного характера неутешительны. Многие больные умирают спустя 5-8 лет после появления первых симптомов от патологий дыхательной системы, которые носят инфекционный характер (например, от воспаления легких) или от образовавшихся в организме злокачественных опухолей. Как правило, больные данным синдромом живут в среднем до 14-15 лет, но бывают редкие исключения, когда при благоприятных условиях жизни пациенты могут дожить до 40 лет.
Профилактических или предупредительных мер заболевания нет по той простой причине, что невозможно оказать влияние на генетику эмбриона в утробе матери.
В статье были рассмотрены характерные проявления синдрома Луи-Бар.
Читайте также:
