
Атаксия фридрейха клинические рекомендации
Атаксия Фридрейха – это наследственное заболевание, которое встречается в неврологической практике крайне редко, менее, чем в 1% случаев. Для того, чтобы оно проявилось, необходимо наличие рецессивного гена у обоих родителей ребенка. Патология впервые дает о себе знать в 10-13-летнем возрасте.
Лечение атаксии Фридрейха – симптоматическое. Ребенок рано становится инвалидом, а средняя продолжительность жизни составляет до 15 лет с момента начала заболевания. Но были зафиксированы случаи, в которых пациенты доживали до 70-80 лет.
Опасность заключается в том, что болеет данной атаксией 1 на 100 тысяч населения, а мутация есть у 1 из 100 человек.
Причины
Тип наследования данного заболевания – аутосомно-рецессивный. Это значит, что оба родителя ребенка должны иметь рецессивный ген, но сами могут не иметь атаксию Фридрейха. Мужчины и женщины болеют одинаково часто.
Происходит мутация в 9 хромосоме. Затем нарушаются процессы оксигенации фермента ЛДГ, снижается синтез медиатора нервной системы – ацетилхолина в спинном мозге, столбе, мозжечке. Нарушается процесс транспорта железа в митохондрии из-за недостаточности белка фратаксина. Происходит снижение глютамина на определенных участках головного мозга.
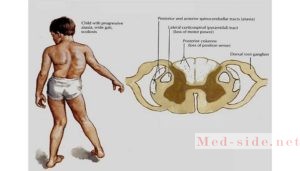
Патанатомически происходит дегенерация спинного мозга, повреждаются боковые канатики. Это – результат действия свободных радикалов. Свободные радикалы также поражают клетки сердца, поджелудочной железы. Антиоксидантные системы организма не успевают обезвредить радикалы.
Симптомы
Первый симптом патологии – неустойчивость походки. Спиноцеребеллярная наследственная или семейная атаксия Фридрейха развивается очень медленно. Пациенту на 4-5 год проявления болезни понадобится инвалидное кресло.
В неврологии выделяют основные симптомы атаксии Фридрейха:
- сенситивно-мозжечковая атаксия,
- нистагм,
- гипотония мышц,
- арефлексия.
После манифестации болезни у пациента выпадает вибрационная и глубокая мышечно-суставная чувствительность. Затем расстройства координации становятся более заметными. Шаткость при ходьбе сопровождается спотыканиями и падениями. Появляется дрожь, тремор в руках, что сопровождается искривлением почерка.
Затем присоединяются нарушения речевого аппарата и слуха (нейросенсорная тугоухость).
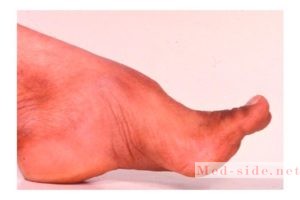
Деформируются пальцы рук и ног, развивается косолапость.
Диагностика
У пациента понижен мышечный тонус, наблюдается снижение сухожильных рефлексов. Исчезает коленный рефлекс, что является ранним признаком атаксии Фридрейха.
Проверить сухожильные рефлексы дома можно самостоятельно. Достаточно ребром ладони легко ударить по сухожилью под коленной чашечкой (на 1,5-2 см ниже). Сначала их проверяют на 1 потом на 2 ноге. При этом пациента просят закинуть ногу на ногу поочередно и расслабиться.
Проверяют устойчивость в позе Ромберга, пациент с атаксией , неустойчив. Пальце-носовая проба также отрицательная, пациент промахивается с закрытыми глазами.
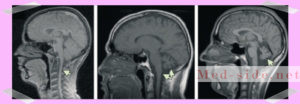
После первичной диагностики у невролога, пациент попадает на МРТ. На снимках визуализируются атрофические процессы в продолговатом мозге и мосте, атрофированный мозжечок. То есть, его размеры уменьшены. Также уменьшается поперечник спинного мозга.
На электромиографии отмечается снижение проводимости по чувствительным волокнам.
Для исследования состояния 9 хромосомы назначается кариотипирование и ПЦР. Дополнительно назначаются консультации у офтальмолога, эндокринолога, кардиолога, делается УЗИ сердца или ЭхоКГ.
Можно определить вероятность атаксии Фридрейха у плода. Проводится ДНК-диагностика ворсин хориона на 8-12 неделе или же для анализа необходим образец амниотической жидкости на 16-24 неделе беременности.
Дифференциальный диагноз
Дифференциальную диагностику проводят с наследственными заболеваниями мозжечка. А именно с такими атаксиями, как:
- наследственная атаксия Пьера-Мари,
- клинические формы оливо-понто-церебеллярной дегенерации,
- болезнь Рефсума,
- болезнь Руси-Левина,
- болезнь Маринеску-Шагрена.
Дополнительно исключают такие патологии, как: опухоль мозжечка, фуникулярный миелоз из-за недостаточности витамина В12, рассеянный склероз, синдром Луи-Барр (атаксия-телеангиоэктазии), сифилис ЦНС.
Клинические рекомендации
Назначают препараты, которые улучшают метаболизм миокарда или сердечной мышцы , тиаминпирофосфат, инозин, триметазидин, 5-гидроксипрофан.
Для улучшения кровообращения в сосудах головного мозга назначают ноотропы и нейропротекторы: энцефабол, гамма-аминомасляную кислоту, пирацетам, меклофеноксат, пиритинол, поливитамины. Уделяют внимание ЛФК и массажам. Иногда проводят хирургическую коррекцию нарушений опорно-двигательного аппарата.
Пиоглитазон и Идебенон – препараты, которые владеют антиоксидантной активностью и содержат коэнзим Q вместе с витамином E. К сожалению, клиническая эффективность этих средств не подтверждена. Об исследованиях можно прочесть здесь.
Поэтому лечение атаксии Фридрейха остается симптоматическим, направленным на поддержание жизни пациента. В случае аритмий необходимо назначить блокаторы кальциевых каналов, антиаритмики. При сахарном диабете – диету с возможным последующим подключением инсулина.
Разрабатывается в Висконсине новая методика лечения, которая предлагает встраивать в ДНК синтетический фактор элонгации. Эксперименты на клетках пациентов с атаксией Фридрейха показали, что Syn-TEF1 , фактор элонгации способен полностью восстановить экспрессию гена FXN.
Патология является неизлечимой. Можно замедлить течение болезни, но в среднем прогноз для жизни составляет 10-15 лет с момента манифестации, то есть клинического проявления атаксии Фридрейха.
Только при отсутствии сахарного диабета и других патологий, особенно со стороны сердечно-сосудистой системы, пациент может дожить до 70-80 лет. Продолжительность жизни с атаксией Фридрейха у женщин благоприятнее, они живут на 30-40% дольше, чем мужчины.
Атаксия Фридрейха (спинноцеребеллярная дегенерация Фридрейха, код по МКБ 10 – G11.1) – вариант ранней мозжечковой атаксии, наследуемой по аутосомно-рецессивному типу, характеризующаяся ранней манифестацией и прогрессирующим течением.
Заболеванию в большей степени подвержены мужчины, с частотой 2-7:100000, у женщин заболевание характеризуется более благоприятным течением, большей продолжительностью жизни. Аутосомно-рецессивный тип наследования подразумевает рождение больного ребенка от клинически здоровых родителей, являющихся носителем патологического гена. Заболеванию не подвергаются представители негроидной расы, представители монголоидной расы страдают крайне редко.
В основе заболевания лежит мутация в длинном плече 9 хромосомы в гене FXN, который отвечает за кодировку белка фратаксина, участвующего в транспорте ионов железа из околомитохондриального пространства. Тем самым данный белок предотвращает появление и токсическое воздействие свободных радикалов (появляющихся в результате высокой концентрации железа) на нейроны, кардиомиоциты, клетки поджелудочной железы, клетки сетчатки и клетки костно-мышечной системы.
При поражении нервной системы в первую очередь дегенерации подвергаются пути Голля, меньше страдают пути Бурдаха, Флексига, Говерса; в процесс вовлекаются спинальные ганглии, спиноцеребеллярные тракты, периферические нервы, структуры мозжечка (их вовлечение в патологический процесс возникает на более поздних этапах развития заболевания). Что касается поражения других систем организма, следует указать на страдание клеток миокарда, поджелудочной железы, сетчатки и костной ткани.
Симптомы

Клинические проявления заболевания зависят от условной формы:
- Классическая болезнь Фридрейха (типичная форма);
- Атипичная форма болезни Фридрейха (при незначительной поломке в 9 хромосоме).
Впервые классическая болезнь Фридрейха (типичная форма) может дать о себе знать в достаточно молодом возрасте, чаще до 25 лет, реже заболевание имеет более медленный темп развития, тогда первые признаки появляются на четвертом десятилетии жизни (атипичная форма).
Манифестирует заболевание чаще с поражения нижних конечностей, нарушений походки и расстройств координации движений (в основе – атаксия, носящая смешанный характер, мозжечково-сенситивный). Пациент отмечает неуверенность при ходьбе, шаткость походки, вплоть до падения. С течением времени может присоединиться нарушение движений верхних конечностей, дрожание в них.
Со временем присоединяются и другие неврологические изменения:
- Слабость в мышцах ног вплоть до парезов и параличей (при этом снижаются, а позже угасают коленные и ахилловы рефлексы, однако могут обнаружиться пирамидные знаки);
- Нарушение глубокой и поверхностной чувствительности;
- Развитие тугоухости, атрофия зрительного нерва;
- Нарушение речи (при длительности заболевания более 5 лет);
- Снижение когнитивных функций.
Соматическими (экстраневральными) проявлениями в клинической картине атаксии Фридрейха являются следующие симптомы:
Учитывая поражение различных структур нервной системы, а также вовлечение в процесс других систем организма, для диагностики принято выделять обязательные признаки, частые признаки, редкие признаки.
К облигатным (обязательным) симптомам болезни относят:
- Дебют заболевания в возрасте до 25 лет.
- Прогрессирующая мозжечково-сенситивная атаксия.
- Прогрессирующее нарушение координации в конечностях.
- Угасание коленных и ахилловых рефлексов.
- Снижение глубокой чувствительности в ногах.
- Дизартрия – толчкообразная, замедленная речь.
К частым, но не обязательным признакам заболевания относят:
- Патологический рефлекс Бабинского.
- Полая стопа.
- Кифосколиоз.
- Слабость, гипотрофия и атрофия мышц конечностей.
Редкие признаки атаксии Фридрейха:
- Атрофия зрительных нервов.
- Нистагм.
- Сенсоневральная тугоухость.
- Постуральный тремор.
- Вертиго.
- Спастичность в конечностях.
- Умеренные когнитивные нарушения.
Врача-клинициста должно насторожить и направить на поиск других заболеваний наличие следующих признаков: ранний манифест заболевания (до 2 лет), выраженные когнитивные нарушения, наличие синдромов экстрапирамидной патологии, сенсо-моторная невропатия, снижение скорости проведения нервного импульса по двигательным волокнам, офтальмоплегия.
Следует помнить, что для атипичной формы заболевания не характерны: тотальная арефлексия, кардиомиопатии, эндокринные нарушения.
Диагностика атаксии Фридрейха

Прежде всего, во время диагностики заболевания, следует оценивать клиническую картину и выявлять обязательные проявления. Также имеется ряд важных инструментальных методик исследования:
- МРТ головного мозга. Позволяет найти атрофию спинного мозга (уменьшение поперечника спинного мозга, увеличивающееся в каудальном направлении – на развернутой стадии). МРТ позволяет визуализировать умеренные атрофические изменения моста, продолговатого мозга и мозжечка.
- На начальных этапах развития заболевания важно проведение нейрофизиологических исследований:
- Электронейромиография: нарушение проведения нервного импульса превалирует в чувствительных волокнах, нежели в двигательных.
- Тест толерантности к глюкозе (исключить СД).
- Рентгенологическое исследование позвоночника и стоп (обнаружение костных деформаций).
- ЭКГ – нарушения проводимости, инверсия з.Т, гипертрофия межжелудочковой перегородки. Следует отметить что нарушения со стороны сердца зачастую предваряют неврологические изменения на несколько лет.
- МСКТ головного мозга. В диагностике заболевания малоэффективна, так как позволяет визуализировать атрофию мозжечка лишь на поздних стадиях. Косвенными признаками заболевания являются: атрофия полушарий, расширение стволовых цистерн, боковых желудочков.
Важно также провести ряд анализов крови: генетическое исследование (ДНК-диагностика – обнаружение мутации в гене FXN методом ПЦР крови) и определение сукцинатдегидрогеназы цитохимическим методом. Важно отметить, что в настоящее время возможна пренатальная диагностика хромосомной мутации.
Дифференциальная диагностика атаксии Фридрейха проводится со следующими заболеваниями:
Лечение
Патогенетического лечения заболевания в настоящее время не разработано, поэтому все меры направлены на устранение симптомов заболевания.
Лечением пациентов с атаксией Фридрейха занимаются врачи нескольких специальностей: кардиологи (к ним часто пациенты обращаются впервые, и нередко эти пациенты наблюдаются с диагнозом ревмокардит несколько лет, до тех пор, пока себя не проявят другие симптомы заболевания), травматологи-ортопеды (хирургическое лечение дефектов стопы, кифосколиоза), эндокринологи (диагностика и лечение сахарного диабета и гормональных дисфункций яичников), реже с данным заболеванием в своей практике сталкиваются ЛОР-врачи и офтальмологи.
Однако чаще, что и логично, пациент наблюдается у невролога, который координирует действия пациента (направляет к узким специалистам для диагностики и коррекции эктраневральных нарушений).
Что касается медикаментозной терапии, лечение носит общеукрепляющий характер:
- Препараты с антиоксидантным действием (этилметилгидроксипиридина сукцинат), при сочетании заболевания с сахарным диабетом – тиоктовая кислота (Берлитион, Тиоктацид, Октолипен); витамины А, Е, поливитамины).
- Средства, улучшающие метаболизм в сердечной мышце и в клетках нервной системы: мельдоний, кокарбоксилаза, рибоксин. Также используют препарат, имеющий структурное сходство с коэнзимом Q10 – идебенон.
- Препараты с ноотропным действием (холина альфосцерат (Церепро, Глиатилин, Ноохолин), пирацетам, аминомасляная кислота).
Немаловажным методом лечения является ЛФК и массаж. Данные методы позволяют пациентам более длительный срок сохранять мышечную силу и поддерживать двигательный режим, также способствуют уменьшению интенсивности болевого синдрома в мышцах конечностей.
Прогноз заболевания
Учитывая отсутствие патогенетического лечения, следует понимать, что заболевание носит неуклонно прогрессирующий характер. Пациенты с диагностированной болезнью в среднем живут 10-15 лет и как правило не более 30 лет с момента начала ее развития, у женщин течение заболевания носит более благоприятный характер – длительность их жизни нередко превышает 20 лет. Летальный исход наступает в результате развития легочной или сердечной недостаточности.
Однако своевременная диагностика и применение вышеуказанных методов лечения, воздействие на сопутствую патологию, недопущение развития осложнений способствуют улучшению качества жизни и ее продлению у пациентов с атаксией Фридрейха.
Симптомы атаксии Фридрейха чаще появляются на первом, втором десятилетии жизни, изредка на третьем и четвертом десятилетии. Появляется неуверенность, пошатывание, спотыкание при ходьбе, частые падения, нарушается почерк из-за тремора, появляется дизартрия, слабость в ногах, нарушается слух. Исчезают сухожильные и надкостничные рефлексы (в первую очередь ахиловые и коленные). Иногда ранним симптомом может быть ревмокардит. Больные не выполняют пяточно-коленную пробу, появляется покачивание в позе Ромберга, которое усиливается при закрывании глаз, расстройства сидения. Симптом Бабинского. Часто нистагм.
Постепенно нарушается глубокая чувствительность, нарастает мышечная атрофия, на начальных этапах более выражена на нижних конечностях, с течением болезни захватывает и верхние. Формируется тотальная арефлексия. Атрофируется зрительный нерв, развивается катаракта, что ведет к слепоте, нарушается функция тазовых органов, развивается деменция.
Течение болезни неуклонно прогрессирующее, при отсутствии адекватного лечения, длительность болезни обычно не превышает 20 лет. Непосредственной причиной смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения. В редких случаях при отсутствии сахарного диабета и сердечных нарушений, больные доживают до 70-80 лет. Прогноз благоприятнее у женщин: более 20 лет с начала заболевания живут 100% женщин и только 63% мужчин.
Диагностика: Компьютерная томография головного мозга, которая остается основной диагностикой атаксий при этом заболевании малоэффективна, т.к. обнаруживает изменения только на поздних стадиях. Удается обнаружить только слабую степень атрофии мозжечка на ранней стадии и атрофию полушарий, расширение стволовых цистерн, боковых желудочков и субарахноидального пространства обоих полушарий на более поздних стадиях. Ранняя диагностика атаксии Фридрейха производится с помощью МР-томографии, которая дает возможность обнаружить атрофию спинного мозга и уменьшение поперечного размера спинного мозга, особенно усиливающееся в каудальном направлении на развернутой стадии, и умерено выраженную атрофию моста, мозжечка и продолговатого мозга. На начальной стадии обязательно проводится электрофизиологическое исследование, при таких исследованиях устанавливается тяжесть поражения чувствительности нервов конечностей. Характерный для данного заболевания электронейромиографический паттерн заключается в отсутствии или значительном снижении амплитуды потенциалов действия чувствительных нервов конечностей, при сравнительно небольшом снижении скорости проведения импульса по двигательным нервам. Для полной диагностики проводят нагрузочные тесты толерантности к глюкозе (для исключения сахарного диабета), рентгеновское исследование позвоночника. На ЭКГ - нарушение ритма, инверсия зубцаТ, изменения проводимости, при эхокарднографии особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки. В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений. Больные длительно наблюдаются у кардиолога или участкового терапевта, чаще всего с диагнозом “ревмокардит”. Для оценки митохондриальных нарушений с помощью цитохимического метода наиболее целесообразным представляется определение активности ряда ферментов-дегидрогеназ лимфоцитов: сукцинатдегидрогеназы (СДГ),
Профилактика атаксии Фридрейха - Особое значение имеет ДНК тестирование на ранней пресимптомной стадии с целью назначения превентивной терапии. Обследуются в первую очередь родственники больного.
Распространенность аутосомно-рецессивной атаксии Фридрейха среди якутского населения PC (Я) составляет 2,8 на 100 тыс. населения. Молекулярно-генетической причиной АФ у якутов является экспансия (GAA) n- повторов в 1 интроне гена FRDA. В улусах вилюйской и центральной Якутии зарегистрированы единичные случаи аутосомно-рецессивной атаксии Фридрейха без накопления в отдельных улусах. Интересен тот факт, что заболевание зарегистрировано у якутского этноса, являющегося представителем азиатской расы, среди которых ранее не было зарегистрировано случаев атаксии Фридрейха.. Это может быть объяснено привнесением в генофонд якутов европейской компоненты, поэтому выяснение причин возникновения и распространения атаксии Фридрейха среди якутов требует дальнейшего изучения.
Похожие темы научных работ по клинической медицине , автор научной работы — Клефортова Инна Игоревна, Корниенко Валерия Александровна, Шестакова Марина Владимировна
Атаксия Фридрейха у больного сахарным диабетом
Клефортова И.И., Корниенко В.А., Шестакова М.В.
ФГУЭндокринологический научный центр, Москва (директор — академик РАН и РАМН И.И. Дедов)
Ключевые слова: атаксия Фридрейха, сахарный диабет, фратаксин
Friedreich's ataxia in a diabetic patient
Klefortova I.I., Kornienko V.A., Shestakova M.V Endocrinological Research Centre, Moscow
Key words: diabetes mellitus, Friedreich’s ataxia, frataxin
Атаксия или болезнь Фридрейха — это наследственное заболевание, связанное с преимущественной дегенерацией проводящих систем спинного мозга и периферических нервных волокон.
Атаксия Фридрейха (АФ) — первая нозологически самостоятельная форма наследственных атаксий, выделенная более 100 лет назад из общей группы локомоторной атаксии Н. Фрид-рейхом в серии своих классических работ 1863 — 1877 гг. Болезнь Фридрейха — самая частая форма наследственных атаксий. В среднем в мире распространенность составляет 2 — 7 случаев заболевания на 100 000 человек, а носителем патологического гена является 1 человек из 120 [1, 2, 3, 4, 9]. Возможно, что частота этого заболевания в популяции выше, — при использовании современных генетических методов диагностики получены несколько иные цифры: заболеваемость — один случай на 29 000 населения, гетерозиготное носительство — один случай на 60 — 90 человек [5].
АФ — это аутосомно-рецессивное заболевание, т.е. больные дети рождаются у пары родителей, оба из которых клинически здоровы, но являются носителями патологического гена. Ген болезни Фридрейха был картирован в центномерной области 9-й хромосомы в локусе 9^13 — q21 (рис. 1) [1, 2, 3, 5, 7, 11, 14].
Рис. 1. Ген фратаксина Х25
общее клеточное железо остается в пределах нормальных значений, а содержание цитозольного железа снижается. Это приводит к активации генов, кодирующих транспортирующее железо — ферменты — ферроксидазу и пермиазу. Таким образом, еще больше усугубляется дисбаланс внутриклеточного железа. Высокая концентрация железа в митохондриях приводит к увеличению количества свободных радикалов, которые обладают повреждающим действием на клетку. Итак, в основе патогенеза АФ лежит нарушение содержания фратаксина, приводящее к митохондриальной дисфункции и обусловленному этим ок-сидантному стрессу [1, 3, 5, 6, 7, 9, 13, 14]. В настоящее время митохондриальный дефект, характерный для этого заболевания, подтвержден не только экспериментальными данными, но и результатами прижизненного исследования у больных скелетных мышц и миокарда при помощи МР-спектрометрии.
Типичным для АФ является начало заболевания на 1 -2-м десятилетиях жизни, отмечаются 2 пика возраста манифестации: в 6-9 лет и 12-15 лет [10]. Симптомы заболевания характеризуются сочетанием типичных неврологических и экстране-вральных проявлений. Заболевание манифестирует обычно появлением неловкости, неуверенности при ходьбе, особенно в темноте, больные начинают пошатываться, часто спотыкаются. Вскоре к атаксии при ходьбе присоединяются дискоор-динация в руках, изменение почерка, слабость в ногах. Уже в самом начале заболевания может отмечаться дизартрия. Ранним и важным дифференциально-диагностическим признаком болезни Фридрейха является исчезновение сухожильных и надкостничных рефлексов. Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько лет опережать манифестацию других симптомов болезни и быть самым ранним проявлением неврологической дисфункции. В развернутой стадии заболевания у больных обычно наблюдается тотальная арефлексия. Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставномышечной и вибрационной) чувствительности. Довольно рано у больных при неврологическом осмотре может быть обнаружен симптом Бабинского, мышечная гипотония. По мере прогрессирования заболевания постепенно нарастают мозжечковая и сенситивная атаксия, слабость и атрофия мышц ног. В поздней стадии болезни часты амиотрофии и расстройства глубокой чувствительности, которые распространяются на руки. Больные перестают самостоятельно ходить и обслуживать себя из-за глубокого распада моторных функций. В ряде случаев наблюдается нистагм, снижение слуха, атрофия зрительных нервов; при длительном течении болезни отмечается нарушение функции тазовых органов, деменция. Среди экстраневральных проявлений АФ необходимо выделить развитие прогрессирующей кардио-миопатии, которая преимущественно носит характер гипертро-
Наследственные нейромышечные заболевания с непереносимостью глюкозы
Синдромы Дополнительные клинические данные
Синдром аносмии гипогонадизма Аносмия, гипогонадизм. Снижение слуха, незаращение губы и неба
Мышечные дистрофии Мышечные дистрофии
Проксимальная миопатия позднего проявления Миопатия, катаракта
Болезнь Гентингтона Хорея, деменция
Болезнь Мачадо Атаксия
Германа синдром Фотомиоклонус, глухота, нефропатия, деменция
Синдром сахарного и несахарного диабета, оптической дистрофии и тугоухости (Вольфрама-Дидмода синдром) Оптическая атрофия, несахарный диабет, глухота, неврологическая симптоматика
Псевдо-Рефсума синдром Мышечная атрофия, атаксия, пигментный ретинит
Атаксия Фридрейха Спиноцеребеллярная дегенерация
Синдром ригидного человека Флюктуирующая мышечная ригидность с болезненными спазмами, характерная ЭМГ, аутоиммунная патология нервной системы и эндокринных желез
Русси-Леви синдром Атаксия, арефлексия, амиотрофия
фической, но в отдельных случаях возможно развитие дилата-ционной кардиомиопатии. Генез кардиомиопатии при АФ связывают с механизмом оксидативного стресса [3, 12, 14]. Пациенты могут предъявлять жалобы на боли в области сердца, учащенное сердцебиение, одышку [1]. Кроме того, нередко возникают аритмии, в том числе фибрилляция предсердий. На ЭКГ изменения — в основном в виде нарушений реполяризации — выявляются у 80% больных, а у 40% при ЭхоКГ определяются признаки гипертрофии миокарда [8]. Кардиальные нарушения являются основной причиной смерти, причем наличие у больных гипертрофической кардиомиопатии вдвое увеличивает риск летального исхода.
К экстраневральным проявлениям болезни Фридрейха относятся эндокринные расстройства (сахарный диабет (СД), ги-погонадизм, гипотиреоз, половой инфантилизм). СД обычно манифестирует уже при длительно текущей АФ (10 лет и более) на фоне инсулинорезистентности, которая в свою очередь связана с наличием мембранных аномалий, приводящих к рецепторным дефектам [15]. Однако в литературе описаны случаи инсулинозависимого СД при АФ, обусловленного потерей ост-ровковых клеток, но без Н^-ассоциации и аутоиммунной деструкции р-клеток. Связь между геном атаксии Фридрейха и геном, определяющим предрасположенность к СД, не выявлена [16].
Для подтверждения диагноза используются методы ДНК-и МРТ-диагностики. Нейроморфологические изменения при АФ в большей мере затрагивают спинной мозг, а не ствол головного мозга или мозжечок [9]. Они заключаются в дегенерации задних столбов спинного мозга и спинно-мозжечковых путей, кортикоспинальных путей, ядер ствола мозга и мозжечка, ножек мозжечка, гибели крупных чувствительных нейронов спинномозговых ганглиев и поражении периферических нервов.
АФ следует дифференцировать с другими наследственными нейромышечными заболеваниями, сочетающимися с СД (табл. 1), а также — от клинически весьма сходной формы наследственной атаксии, вызываемой дефицитом витамина Е, и близкого к ней синдрома Бассена-Корнцвейга. Для диффе-
Представляем клинический случай пациентки с атаксией Фридрейха.
Пациентка родилась от второй нормально протекающей беременности, роды — в срок, при рождении масса тела — 3650 г, длина — 52 см. До 7 лет росла и развивалась соответственно возрасту, от сверстников не отставала. Перенесенные заболевания: краснуха в возрасте 5 лет. Старший брат (сводный, разные отцы) здоров (36 лет), имеет двоих здоровых детей (12 лет и 1 год 3 месяца). Наследственность по СД не отягощена, по АГ у бабушки по материнской линии.
При поступлении предъявляла жалобы на колебания уровня глюкозы крови от 3,0 до 20,0 ммоль/л, гипогликемии в вечерние часы 1-2 раза в неделю, повышение АД — до 140/90 мм рт.ст.
Из анамнеза известно, что СД впервые выявлен в 1990 г. в возрасте 7 лет на фоне выраженного снижения веса. Дебют заболевания — с кетоацидоза. Сразу назначена инсулинотерапия в фиксированных дозах, уровень гликемии при редких измерениях (приблизительно 1-2 раза в год) — в пределах 11,0-13,0 ммоль/л в течение последующих 4-х лет. Через 2 года от начала заболевания (в 9 лет) резко снизилась острота зрения.
Данные лабораторного исследования: в общем анализе крови и мочи — без особенностей. В биохимическом анализе крови обращает на себя внимание уровень креатинина 84 мкмоль/л, уровень мочевины — в пределах референсных значений, СКФ (MDRD) — 76 мл/мин/1,73 м2. В анализе утренней мочи на микроальбуминурию уровень альбумина — 1,0 мг/л (норма 0 — 20,0). НЬА1с — 7,8%; С-пептид 0 и 120 минута — Не можете найти то, что вам нужно? Попробуйте сервис подбора литературы.
Больной проводилось лечение: стол № 9, с ограничением легкоусваиваемых углеводов; инсулинотерапия: Лантус 18 Ед в 22.00, Хумалог 12-10-10 Ед перед основными приемами пищи; Конкор 2,5 мг 2 раза в день; Кораксан 5 мг утром; Эль-кар 20% 1 ч.л. 3 раза в день, курсами по 2 месяца 3 раза в год; Коэнзим Q10 (60 мг) по 1 капсуле 3 раза в день, курсами по 2 месяца 3 раза в год; Трентал (100 мг) по 1 таб. 3 раза в день
Рис. 4Б. УЗИ орбит (OS) у пациентки Н., 25 лет
после еды 2 месяца; Церебролизин 5,0 мл в/в струйно или в/м через день, № 10-15; Дюфастон 10 мг по 1 таб. 2 раза в день с 15 по 26 д.м.ц.
Рекомендации: консультация в Институте неврологии для решения вопроса о госпитализации; экстракция катаракты; изготовление сложной ортопедической обуви по ИПР.
Прогноз благоприятнее у женщин: более 20 лет с начала заболевания живут 100% женщин и только 63% мужчин; 50% больных не доживает до 35 лет. Заболевание неуклонно прогрессирует и приводит к глубокой инвалидизации, но темпы прогрессирования различны: иногда больные в течение нескольких лет перестают ходить, в других случаях сохраняют возможность передвижения и самообслуживания достаточно долго. Нарушение функции тазовых органов иногда появляется у больных с длительно текущим заболеванием. Интеллект у пациентов с данным заболеванием в большинстве случаев не страдает. Причиной смерти зачастую является прогрессирующая кардио-миопатия.
2. Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК-диагностика и медико-генетическое консультирование в неврологии. - М.: Медицинское информационное агентство, 2002. - 591 с.
3. Chakravarty A. Friedreich's ataxia - yesterday, today and tomorrow// Neurol. India. - 2003. - 51. - Р. 176-182.
4. Civenni G., Bezzi P., Trotti D. et al. Inhibitory effect of the neuroprotec-tive agent idebenone on arachidonic acid metabolism in astrocy-tes// Eur. J. Pharmacol. - 1999. - 370. - Р. 161-167.
5. Delatycki M.B., Williamson R., Forrest S.M. Friedreich ataxia: an overview // J. Med. Genet. - 2000. - 37. - P. 1 -8.
6. De Michele G., Coppola G., Cocozza S., Filla A. A pathogenetic classification of hereditary ataxias: Is the time ripe? // J. Neurol. - 2004. - 251. - P. 913-922.
7. DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases // New Engl. J. Med. - 2003. - 348. - P. 2656-2668.
8. Emond M., Lepage G., Vanasse M., Pandolfo M. Increased levels of plasma ma-londialdehyde in Friedreich ataxia // Neurology. - 2000. - 55. - P. 17521753.
9. Filla A., Cocozza S., De Michele G. Friedreich's ataxia: from the patient to the gene // Neurol. Sci. - 2001. - 22. - P. S21-S25.
10. Filla A., Michele G., Caruso G. et ol. // Amer. J. Hum. Gepat. - 1996., V. 59. -P. 554-560.
11. Filla A., Moss A.J. Idebenone for treatment of Friedreich's ataxia? //Ne- 14.
urology. - 2003. - 60. - P. 1569-1570.
12. Lang D. Cardiac hypertrophy and oxidative stress: a leap of faith or stark 15. reality? // Heart. - 2002. - 87. - P. 316-318.
13. Schon E.A., Manfredi G. Neuronal degeneration and mitochondrial dys- 16. function // J. Clin. Invest. - 2003. - 1 11. - P. 303-312.
Sherer T., Greenamyre J.T. A therapeutic target and biomarker in Frie dreich's ataxia // Neurology. - 2000. - 55. - P. 1600-1601.
Fantus I.G., Senni M.N., Andermann E.J. // Clin. Endocrinol. Metab. -1993. - Jan., 76(1). - P. 60-63.
Schoenle E.J., Boltshauser B.J., Balkkeskov S. et al. // Diabetologia. -1989. - Jan.; 32(6). - P. 378-381.
Клефортова Инна Игоревна к.м.н., старший научный сотрудник, ФГУ Эндокринологический научный центр, Москва
Корниенко Валерия Александровна клинический ординатор, ФГУ Эндокринологический научный центр, Москва
Шестакова Марина Владимировна д.м.н., профессор, директор Института диабета, ФГУ Эндокринологический научный центр, Москва
Читайте также:
