
Болезнь гентингтона что это такое простыми словами

Болезнь Гентингтона является генетической болезнью с аутосомно-доминантным типом наследования, которая проявляет себя во взрослом возрасте. Впервые заболевание было описано в 1872-м году Джорджем Гентингтоном и названо в его честь.
- Описание заболевания
- Причины развития заболевания
- Патогенез
- Стадии заболевания
- Факторы риска
- Формы патологии
- Симптомы и первые признаки
- Диагностика
- Методы лечения
- Прогноз у детей и взрослых
- Что нужно запомнить?
Описание заболевания
Морфологическая картина связана с поражением corpus striatum (стриатума) – структуры конечного мозга, состоящей из крупных нейронов. 
Данный отдел отвечает за регуляцию мышечного тонуса, поведенческие реакции и формирование условных рефлексов.
При болезни Гентингтона наблюдается стремительная гибель нейронов стриатума, приводящая к его атрофии, а также появление зон глиоза. В МКБ-10 болезни Гентингтона присвоен код G10.
Заболевание является редким и мало изученным. В США частота встречаемости составляет 4,1-8,4 случаев на 100000 человек, в странах Европы – 1,63-9,95 случаев на 100000 населения. Тем не менее, эти данные весьма приблизительны. В литературе не описано случаев начала заболевания в возрасте до 10-ти и после 70-ти лет. Интересно, что у жителей Венесуэлы средний возраст начала болезни 34—35 лет, а у американцев – 37—47, у жителей Канады – 40—36 лет. Большинство исследований показало, что средний возраст появления клинических симптомов составляет 35—44 года.
Причины развития заболевания
Развитие болезни изначально связано с синтезом мутантного белка хантингтина Htt. Все симптомы болезни Гентингтона возникают вследствие дисфункции и постепенной потери нейронов в стриатуме, коре и других структурах мозга. Существует несколько факторов, которые могут влиять на клеточную гибель. К ним относятся:
- Экзитоксичность – активация нейротрансмиттеров, которая приводит к повреждению нейронов.
- Оксидативный стресс – результат присутствия свободных радикалов в большом количестве.
- Нарушения нейрометаболизма. Исследования с помощью ядерно-магнитной резонансной спектроскопии показала высокие уровни молочной кислоты в базальных ганглиях и затылочной части коры головного мозга у пациентов с болезнью Гентингтона.
Апоптоз, или запрограммированная гибель клетки, является результатом вышеперечисленных процессов.
Патогенез
Ген HTT, кодирующий синтез белка хантингтина (Htt), локализован на коротком плече 4-й хромосомы (4р16.3) и есть у всех людей. Он содержит участок с кодоном цистеин-аденозин-гуанин (ЦАГ), который кодирует синтез глутамина. Генетическая причина болезни связана с ростом повторов кодона ЦАГ. Количество повторов триплета ЦАГ более чем 36 раз приводит к синтезу мутантного белка, который становится токсичным и серьезно повреждает клетки.
Функция белка Htt до сих пор неизвестна медицинской науке. Белок располагается в цитоплазме, принимает участие в передаче сигнала в клетке и предотвращает ее запрограммированную гибель. N-концевые фрагменты мутантного хантингтина (mHtt) формируют включения в клеточных ядрах нейронов, провоцируя таким образом их повреждение. Исследования культур нейронов стриатума с включениями показали, что данный феномен встречается редко и чаще подобные изменения находят в нейронах коры головного мозга.
Недавнее исследование проводилось на протяжении 12 месяцев с вовлечением трех групп пациентов, две из которых включали больных с мутантным геном Htt. В исследование были включены 116 человек-носителей мутантного гена без клинических проявлений заболевания; 114 — с ранними признаками болезни и 115 из группы контроля (без мутантного гена). У пациентов из первых двух групп достоверно чаще наблюдались признаки атрофии вещества мозга (как локальных участков, так и обширных зон поражения), замедление мыслительных процессов, ухудшение моторных функций, появление атипичных глазодвигательных реакций и уменьшение общего объема мозга по сравнению с группой контроля.
Стадии заболевания
В зависимости от объема поражения стриатума, гибели нейронов и наличия глиоза выделяют несколько стадий болезни:
Стадия 0 подразумевает отсутствие атрофии. На этом этапе не определяется признаков поражения вещества мозга при наличии клинических проявлений и положительной семейной истории.
Стадия 1 – микроскопически подтвержденные нейропатологические изменения, без признаков атрофии.
Стадия 2 – стриатум с атрофией, хвостатое ядро структурно не изменено, имеет выпуклую форму.
Стадия 3 – атрофия резко выражена, хвостатое ядро заметно уплощено.
Стадия 4 – тяжелая атрофия, хвостатое ядро выглядит запавшим.
На рисунке 1 показана область поражения головного мозга при болезни Гентингтона.
Факторы риска
Выделяют три главных фактора, значительно повышающих не только риск развития болезни Гентингтона, но и ранее ее начало. К ним относятся:
- Количество повторов в триплете ЦАГ гена, кодирующего синтез хантингтина.
- Нестабильность триплета ЦАГ.
- Влияние генов-модификаторов.
Среди всех потенциально значимых факторов наиболее важным является количество повторов в мутантном аллеле, что ассоциируется со стремительным прогрессированием болезни. Количество повторов ЦАГ обратно пропорционально коррелирует с возрастом появления симптомов. Кроме того, высокое количество повторов триплета ЦАГ сочетается с более высоким риском развития инвалидности и смертности.
Формы патологии
Отдельно выделяют ювенильную форму заболевания (Westphal variant), которая развивается у 5-10% пациентов с мутантным геном. Большинство пациентов с ювенильной формой болезни получают патологический ген от отца, в то время как пациенты с дебютом болезни после 20-ти лет – от матери. В большинстве случаев, клиника развивается у молодых людей в возрасте до двадцати лет. Наиболее часто встречаются:
- дистония;
- деменция;
- эпилепсия;
- хорея.
Симптомы и первые признаки
Клинические проявления заболевания включают двигательные нарушения (чаще всего хорею), когнитивные расстройства и нарушения поведения. К ранним признакам болезни относят:
- Появление нетипичных движений глазных яблок.
- Дизартрию.
- Дисфагию.
- Утрату кратковременной памяти.
Тяжесть симптомов варьирует и связана со степенью поражения мозга. Например, у пациентов со стадией 1-2 проявляются когнитивные расстройства, если стадия 3-4 присоединяются также двигательные нарушения. Однако даже на ранних этапах может быть сочетание всех симптомов болезни.
Изменения поведения, которые проявляются высокой раздражительностью, неопрятностью и потерей интереса к окружающему миру считаются предвестниками деменции. Позже присоединяются нарушения памяти, интеллектуальный дефицит, трудности с восприятием новой информации. Депрессия также является симптомом болезни Гентингтона, поэтому повышается риск суицида. У пациентов отмечаются расстройства психики, проявляющиеся эпизодами различных маний, биполярным расстройством, психозом, обсессивно-компульсивными расстройствами, нарушениями сна, сексуальными девиациями, признаками изменения личности.
Диагностика
Диагностика базируется на клинической картине и данных анамнеза пациента. Ни компьютерная (КТ), ни магнитно-резонансная томография (МРТ) не являются эффективными способами диагностики болезни Гентингтона. Тем не менее, измерение бикаудального диаметра (расстояния между головками двух хвостатых ядер) с помощью КТ или МРТ считается надежным маркером болезни.
Генетическое тестирование основано на подсчете количества повторов триплета ЦАГ в каждом аллеле, однако исследование используется не часто, если присутствует типичная клиническая картина и отягощенный семейный анамнез.
На сегодняшний день опубликованы результаты исследования, показавшие, что уровень белка Htt в слюне у пациентов с болезнью Гентингтона существенно выше (0,775 нг/мл) по сравнению с уровнем Htt слюны здоровых людей (0,359 нг/мл). Исследования продолжаются и, если достигнут высокого уровня доказательности, станет возможным определение Htt в слюне, как раннего маркера болезни Гентингтона.
Всем пациентам из группы риска рекомендуется:
- консультация генетика;
- невролога;
- психиатра.
Если генетическое тестирование показало отрицательный результат, рекомендовано обследование на заболевания щитовидной железы, антифосфолипидный синдром, системную красную волчанку, нейроакантоцитоз, болезнь Вильсона, дендаторубро-паллидолуизальная атрофия.
Методы лечения
Эффективной терапии не разработано и все способы лечения являются экспериментальными. Наиболее известными являются:
- Аблятивные хирургические процедуры;
- Трансплантация фетальных клеток.
Достаточного количества данных, подтверждающих эффективность этих методов, нет.
Методы симптоматической терапии зависят от присутствия доминирующего синдрома:
- если выражена хорея, применяются бензодиазепины – клоназепам и диазепам, вальпроевая кислота, резерпин и нейролептики;
- у пациентов с депрессией применяют ингибиторы обратного захвата серотонина в качестве препаратов первой линии.
- при галлюцинациях используют антипсихотические средства.
- при высокой раздражительности используют антидепрессанты.
Не смотря на то факт, что не существует терапии, которая могла бы устранить симптомы болезни, симптоматическое лечение улучшает качество жизни и предупреждает развитие осложнений. Не рекомендуется назначение множества препаратов одновременно, так как у пациентов чаще развиваются побочные эффекты. Лечение сводится к назначению препаратов для облегчения двигательных расстройств и препаратов для коррекции психоэмоциональной сферы.
Прогноз у детей и взрослых
Болезнь Гентингтона является прогрессирующим заболеванием нервной системы, которое приводит к инвалидности и смерти, преимущественно, от присоединившихся заболеваний. Средний возраст смерти составляет 51-57 лет, однако границы могут быть больше. Продолжительность болезни различная, в среднем, от начала заболевания до терминальной стадии проходит около 19 лет. Самыми частыми причинами смерти являются пневмония и сердечно-сосудистые заболевания.
Посмотрите полезное видео о пациентах с болезнью Гентингтона.
Что нужно запомнить?
- Болезнь Гентингтона – это тяжелое прогрессирующее генетическое заболевание с аутосомно-доминантным типом наследования, связанное с синтезом дефектного белка хантингтина (Htt);
- Молекулярная причина болезни связана с резким увеличением количества повторов триплета цистеин-аденозин-гуанин в гене, кодирующем синтез белка Htt;
- Дефектный белок Htt вызывает повреждение стриатума, базальных ганглиев и коры головного мозга вследствие формирования включений в клеточных ядрах нейронов;
- В зависимости от объема поражения мозга, выделяют пять стадий заболевания;
- Главным фактором, который ассоциируется с прогрессированием болезни, является количество повторов в мутантном аллеле гена Htt;
- Различают две формы болезни: ювенильную, с дебютом до 20-ти лет, и старшего возраста – с началом после 20-ти лет;
- При болезни Гентингтона основными симптомами являются хорея, когнитивные расстройства и нарушения поведения;
- В качестве диагностики используют дорогостоящее генетическое исследование, основанное на подсчете количества триплетов ЦАГ в каждом аллеле гена HTT. Ведутся исследования по определению белка Htt в слюне, как раннего маркера болезни;
- Специфического лечения не существует, используют лишь паллиативные методы, улучшающие качество жизни пациента и членов его семьи;
- Прогноз неблагоприятный, так как заболевание неуклонно прогрессирует.
Литература
- Loy CT, McCusker EA. Is a motor criterion essential for the diagnosis of clinical huntington disease?. PLoS Curr. Apr 11 2013. 5.
- Stober T, Wussow W, Schimrigk K. Bicaudate diameter—the most specific and simple CT parameter in the diagnosis of Huntington’s disease. Neuroradiology. 1984. 26(1):25-8.
- Huntington G. On chorea. Med Surg Report. 26:320.
- Folstein SE. Huntington’s Disease: A Disorder of Families. The Johns Hopkins University Press.
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. Mar 9 2004. 101(10):3498-503.
- Wexler NS, Young AB, Tanzi RE, Travers H, Starosta-Rubinstein S, Penney JB, et al. Homozygotes for Huntington’s disease. Nature. Mar 12-18 1987. 326(6109):194-7.
- Ho A, Hocaoglu M. Impact of Huntington’s across the entire disease spectrum: the phases and stages of disease from the patient perspective. Clin Genet. Sep 2011. 80(3):235-239.
- Loy CT, McCusker EA. Is a motor criterion essential for the diagnosis of clinical huntington disease?. PLoS Curr. Apr 11 2013. 5.
- Huntington Study Group, Frank S, Testa CM, Stamler D, Kayson E, Davis C, et al. Effect of Deutetrabenazine on Chorea Among Patients With Huntington Disease: A Randomized Clinical Trial. JAMA. 2016 Jul 5. 316 (1):40-50.
Исследователи выделяют несколько форм хорей, одна из самых сложных – хорея Гентингтона. Заболевание встречается относительно редко, обусловлено наследственными причинами. Сопровождается двигательными, эмоциональными нарушениями и расстройством высших психических функций. Носит дегенеративный характер.

Хорея
Движения больного напоминают хаотические танцевальные действия. Обусловлены они непроизвольными сокращениями мышц различных частей тела.
Этиология заболеваний, связанных с хореическими гиперкинезами, различна. В связи с этим принято выделять 3 вида хореи.
К данному виду относится хорея Гентингтона (названа по фамилии обнаружившего ее исследователя George Huntington). Это редкое заболевание, характеризующееся медленным прогрессирующим течением. Встречается в среднем в 5 случаях на 100 000 человек. Проявляется преимущественно в возрасте от 30 до 40 лет и старше, в большей степени у мужчин. Наиболее ярко выражены двигательные нарушения, расстройства психики, интеллекта, памяти.

На первых этапах ее признаки найти удается не всегда. Со временем наблюдается подергивание мышц лица, человек начинает совершать непроизвольные глотательные, сосательные движения. Отмечается нескоординированные движения глаз, нистагм. Поражаются руки, появляется симптом играющих на пианино пальцев. Изменяется походка, становится невозможным сохранить положение тела. Болезнь затрагивает все больше групп мышц, постепенно они не только начинают хаотически двигаться, но также становятся ригидными. На поздних этапах человек теряет способность самостоятельно передвигаться.
Со временем ухудшается внимание, память. Человек теряет способность анализировать, логически мыслить, решать даже простейшие задачи, сосредотачиваться. По мере прогрессирования заболевания развивается деменция.
На фоне невозможности регулирования движений, осознания тяжести происходящих в организме процессов, имеющих патологический характер, появляется апатия, склонность к депрессии, повышенная тревожность. Болезнь Гентингтона характеризуется уплощением эмоций. Снижение критического мышления ведет к приступам ярости, повышению агрессии. В ряде случаев проявляется склонность к суициду.
Следствием поражения мышц лица, носоглотки является нарушение речи. Она становится смазанной, невнятной. Снижение интеллекта и памяти приводит к уменьшению словарного запаса.
Синдром хореи встречается в составе других наследственных болезней, связанных с поражением экстрапирамидной системы. Это редко встречающийся (1:300 000) синдром Леша-Нихена, связанный с нарушением содержания мочевой кислоты.
Другой патологией считается врожденная болезнь Вестфаля-Вильсона, в основе которой лежит нарушение усвоения меди.
Хорея возникает в результате травм, сосудистых заболеваний, воздействия бактерий, вирусов, интоксикации, дисбаланса обмена веществ.
К вторичным заболеваниям относят ревматическую (малую) хорею. Ее появлению предшествует обострение ревматизма или стрептококковые инфекции. Болезнь чаще возникает у детей и в возрасте до 20 лет, больше у девочек.
На начальных этапах у ребенка возникают гримасы, нарушаются движения глаз, появляются простые неконтролируемые жесты. В дальнейшем поражаются другие группы мышц. При сильно выраженных проявлениях гиперкинеза ребенок теряет возможность ходить, не может сам кушать, одеваться. Нарушается речь, интеллект, дыхание. Уменьшение яркости симптомов при правильном лечении проходит в основном через шесть месяцев. Возможно появление рецидивов.
Повторное появление возможно, в частности, при беременности. Связывают ее с образованием антител к фосфолипидам. Признаки недуга возникают на третьем месяце. Проявляется гиперкинезами, слабостью мышц, интоксикацией. Протекает болезнь обычно очень тяжело. Является основанием для того, чтобы прервать беременность.
Механизм развития
Вне зависимости от вида хореи наблюдается поражение продолговатого отдела головного мозга, который отвечает за рефлекторную двигательную активность, а также принимает участие в выполнении произвольных движений.
Продолговатый мозг выполняет ряд функций:
- Рефлекторные. Регулирует дыхание, тонус мышц, глазодвигательные, защитные рефлексы. К последним, в частности, относится глотание, моргание, кашель, чихание.
- Проводниковые. Здесь проходят нервные импульсы к головному мозгу от спинного и обратно.
- Интегративная. Продолговатый мозг отвечает за выполнение некоторых сложных функций, например, регуляцию положения глаз во время движения головой.
Поражение участков продолговатого мозга ведет к нарушению этих функций.
Причины
К заболеванию ведет целый ряд причин.
У хореи Хантингтона – генетическая основа. Наследуется она по аутосомно-доминантному признаку. Участок короткого плеча четвертой хромосомы, ответственный за выработку белка гентингтина, в норме имеет 36 повторов тринуклеотидов цитозинаденина-гуанина. В некоторых случаях патологические изменения ведут к увеличению повторов до 121. Образующаяся неправильная структура белка ведет к гибели нейронов мозга. Исследователи отметили закономерность – чем больше повторов, тем раньше возникнет хорея. При изменении повторов до 39 говорят о сомнительном статусе.

К развитию вторичной хореи приводит стрептококковая инфекция, ревматизм, нарушения в работе иммунной системы, черепно-мозговые травмы, сосудистые нарушения, интоксикация. Считается, что причины заболевания кроются в нарушениях обмена веществ, системной красной волчанке.
Симптомы
Возраст первого проявления признаков хореи зависит от формы заболевания. При ревматической хорее и ювенильной форме симптомы возникают в детстве и юношестве. При хорее Гентингтона они появляются после 30 лет.

Отмечается появление гримас, больной непроизвольно высовывает язык, хмурится, издает странные звуки, шмыгает носом. Чуть позже начинает кивать головой, махать руками, покачиваться. Человек теряет способность удерживать взгляд на каком-либо предмете, не может сохранять позу. В начале развития патологии он пытается удержаться от непроизвольных действий, но в дальнейшем ему это удается все с большим трудом.
Признак, который обращает на себя внимание достаточно рано, – нистагм.
С течением времени проявления гиперкинеза ослабевают, повышается тонус. При этом чувствительность не страдает.
Возникают психические и эмоциональные нарушения. Больные становятся вспыльчивыми, эмоциональными, порой – агрессивными. На поздних этапах возможны галлюцинации, мысли о суициде. Нарушается память, критическое мышление, снижается внимание. Речь становится невнятной и неразборчивой.
Симптоматика ювенильной формы дополняется эпилептическими приступами.
Диагностика
Наиболее достоверным способом считается метод полимеразной цепной реакции. Для его выполнения берут из вены кровь и в ходе исследования подсчитывают количество повторов тринуклеотидов цитозинаденина-гуанина. Проводится ДНК-диагностика.
Среди других методов, позволяющих увидеть нарушения продолговатого мозга и признаки инфекций:
- магнтитно-резонансная томография;
- анализ крови;
- электроэнцефалография;
- электромиография.

На осмотре врач использует провокационные пробы. В ходе одной из них пациента просят встать так, чтобы ноги были вместе, а руки разведены в стороны, при этом ладони должны быть обращены вниз. Больного просят открыть, а затем закрыть глаза.
Другая проба – попросить больного лечь и вытянуть руки.

Общие сведения
Хорея, что за болезнь? Хорея (или хореический гиперкинез) — это непрерывные, быстрые, хаотичные движения. Гиперкинез вовлекает дистальные отделы рук и ног, мимические мышцы, мышцы гортани и туловища. Движения эти насильственные и напоминают гримасничанье, ужимки и кривляние. Гиперкинезы связаны с поражением структур головного мозга, объединяемых в экстрапирамидную систему. Эти двигательные расстройства значительно ограничивают возможности больного, приводя к социальной изоляции. Частыми формами хореи являются хорея Гентингтона и ревматическая хорея (синоним пляска Святого Витта и хорея Сиденгама), которые имеют различное происхождение и прогнозы.
Также к гиперкинезам, связанным с поражением экстрапирамидной системы, относятся кривошея, тремор, дистония, атетоз, тики, миоклония. Все эти состояния характеризуются гипотонико-гиперкинетическим синдромом — то есть сочетают гиперкинезы (непроизвольные, насильственные движения) и сниженный тонус мышц. Функция экстрапирамидной нервной системы состоит в создании условий для движений (распределение мышечного тонуса и подготовка мышц к движению) обеспечении позы и выполнении стереотипных рефлекторных движений. Все эти функции нарушаются при нарушении функции экстрапирамидной системы.
Патогенез
При данном заболевании имеет место преимущественное поражение базальных ганглиев головного мозга, которые имеют большое значение в контроле движения и поведения. Также снижается уровень нейромедиаторов (гамма-аминомасляной кислоты и субстанции Р). Данные нарушения возникают вследствие генных мутаций.
Ген хореи Гентингтона локализуется на четвертой хромосоме. Ген кодирует белок хантингтин (гентингтин), который необходим для развития и выживания клетки. В норме ген содержит цепочку из нуклеотидов цитозин-аденин-гуанин (CAG), которая повторяется. Число таких повторов в гене в норме от 8 до 35, а в мутагенном гене увеличивается количество повторов и это обуславливает болезнь Гентингтона. Поскольку триплет CAG кодирует аминокислоту глутамин, при этом заболевании глутамин накапливается в избытке и оказывает повреждающее действие. Чем больше CAG-копий, тем раньше развивается заболевание и больше выражены симптомы.
Классификация
Экстрапирамидные гиперкинезы бывают:
- Первичные (при различных нейродегенеративных заболеваниях, например, болезнь Гентингтона, нейроакантоцитоз, спиноцеребеллярная атаксия).
- Вторичные (симптом при аутоиммунных, сосудистых и метаболических заболеваниях, интоксикации тяжелыми металлами, энцефалитах различной этиологии).
По распространенности хореические гиперкинезы делятся на:
- Фокальные.
- Односторонние.
- Генерализованные.
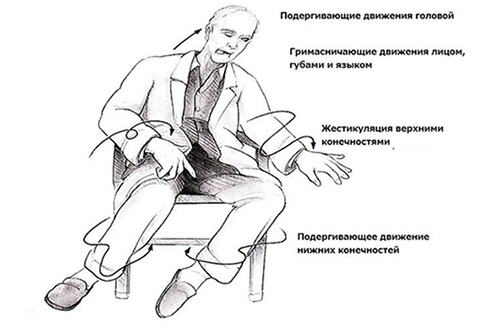
Уже на ранней стадии болезнь Гентингтона помимо двигательных расстройств появляется выраженное нарушение речи (замедленная аритмичная речь), расстройство глотания появляется на поздних стадиях и становится причиной аспирации, асфиксии и пневмонии. Изменения со стороны психической сферы представлены нарушением памяти, внимания, фобическими расстройствами и депрессией с суицидальными попытками.
Среди вторичных форм выделяется малая хорея (синоним хорея Сиденгама), которая является осложнением ревматизма. Это общепризнанный вариант ревматического поражения нервной системы, поэтому ревматическая хорея называется еще нейроревматизм. Первые описания эпидемии ревматической хореи сделаны в 1418 г., а полная характеристика болезни дана Сиденгамом через 200 лет. В 1831 г. Брайтом установлена связь малой хореи с ревматической лихорадкой и описаны тяжелые формы псевдопаралитической малой хореи.
В последние годы благодаря противоревматическому лечению хорея Сиденгама встречается очень редко, да и сам ревматизм ревматологи рассматривают как заболевание, теряющее свою актуальность. В начале 80-х годов нейроревматизм (пляска святого Витта) диагностировался у 36% детей с ревматизмом, а в настоящее время у 16% пациентов. При этом преобладает гемихорея (гиперкинезы одной стороны) и мышечная гипотония (умеренная и выраженная), нарушения координации, почерка и речи, повышение сухожильных рефлексов и эмоциональная лабильность.
Причины хореи Хантингтона
Причина данного заболевания — генетический дефект в четвертой хромосоме. Генетические причины хореи Хантингтона заключаются в изменении 4-й хромосомы, в которой увеличивается количество повторов одного из фрагментов ДНК, который кодирует белок гентингтин. Чем больше количество повторов, тем в более раннем возрасте развивается заболевание. Эти генетические причины и связанное с ними нарушение образования белка за счет полиглутаминовых повторов предопределяют гибель нейронов полосатого тела и хвостатого ядра.
Если говорить о вторичных формах хореи, то причинами их в детском возрасте могут быть ревматизм, системная красная волчанка, поражение сосудов (сосудистая хорея), антифосфолипидный синдром. У пожилых хорея может быть вызвана заболеваниями печени, инсультом, полицитемией. Хорея является самым частым постинсультным гиперкинезом и наблюдается у 1,3% пациентов с острым нарушением мозгового кровообращения. Хореический гиперкинез появляется остро в первые дни инсульта. Он чаще представлен гемихореей, а в случае двустороннего поражения сосудов — генерализованный. У большинства больных гиперкинез сочетается с мышечной слабостью этой же стороны.
Причиной сосудистой хореи является ишемическое или геморрагическое поражение таламуса, субталамического ядра и лентиформных ядер. Причиной сосудистой хореи также может стать поражение теменных, лобных или височных долей.
Симптомы хореи Гентингтона
Симптомы включают различные виды патологических движений. Они проявляются сначала в дистальных отделах конечностей, а затем становятся генерализованными, что нарушает выполнение обычных движений. Сначала у больных появляется неповоротливость, неуклюжесть, плохая координация движений и ухудшается почерк. Быстрые и беспорядочные движения в разных группах мышц связаны со снижением тормозного влияния головного мозга на двигательные нервы.
В 60% случаев до возникновения двигательных расстройств появляются психопатические нарушения.

Болезнь Гентингтона: симптомы и признаки, фото
Эмоциональные нарушения и повышенная возбудимость проявляются тем, что у больного появляются немотивированные приступы паники, ярости, тревоги. Также может развиться гиперсексуальность. Отмечаются нарушения сна.
Слабоумие развивается на поздних стадиях заболевания. Иногда развитие деменции приостанавливается, и оно далеко не всегда становится тотальным. Поэтому можно сказать, что слабоумие при хорее Гентингтона относительно доброкачественное. Некоторые больные могут долго выполнять привычную работу и оставаться вне психиатрической лечебницы. Расстройства интеллекта проявляются в снижении способности к мышлению, ослаблении внимания и отсутствии критики к своему поведению. Если рассматривать другие признаки, то нужно отметить высокую подверженность больных к депрессивным расстройствам и наклонность к суициду. Могут появиться бред и навязчивые состояния. Могут наблюдаться эпилептические приступы — чаще всего это встречается у больных с началом заболевания в молодом возрасте. Болезнь Хантингтона может сочетаться с сахарным диабетом. Поэтому у таких больных часто выявляют симптомы диабета: повышенная жажда, увеличение количества мочи, сухость во рту.
В зависимости от преобладания в клинике того или иного симптома, выделяют несколько клинических форм заболевания:
- Гиперкинетическая. Преобладает непроизвольное движение мускулатуры, усиливающееся при волнении. При ходьбе больные размахивают руками, пританцовывают, раскачиваются, гримасничают, издают посторонние звуки при разговоре. Гиперкинезы исчезают во сне. Со течением времени они нарастают и больные не способны обслуживать себя.
- Психическая форма. У больных на первый план выступают апатия, снижение критики, ухудшение памяти, возбуждение, бредовые идеи, возникают галлюцинации (слуховые и зрительные). Постепенно развивается деменция.
- Акинетико-ригидная форма. Эта форма проявляется мышечной ригидностью и контрактурами. Бывают приступы эпилепсии, атетоз (движения в дистальных отделах рук и ног), глазодвигательные нарушения, атаксия и дистония. Отмечаются также нарушение умственного развития.
Важное место занимают системные проявления ревматизма: одышка, сердцебиение в покое, увеличение печени, боли в суставах, наличие кольцевидной эритемы на коже. Поражения сердца (эндо— и миокардит) выявляется на ЭХО кардиографии: расширение отделов сердца, изменения клапанов сердца, дисфункция створок клапанов. То есть сочетается неврологическая симптоматика и признаки эндомиокардита.
Анализы и диагностика
- Синдром Хантингтона, как наследственное нейродегенеративное заболевание, нуждается в проведении медико-генетических обследований. Проводится генетическое тестирование — прямая ДНК-диагностика фрагментным анализом. Это дает возможность выявить носительство патологического гена у будущих родителей и дать точный прогноз в отношении здоровья потомства.
- МРТ и КТ выявляют атрофию хвостатого ядра, атрофию коры головного мозга (больше в лобной доле), но при генетически подтверждённом диагнозе эти обследования не имеют диагностического значения.
При исследовании больных с ревматической хореей выявляют:
- Лейкоцитоз.
- Нейтрофилез.
- Ускорение СОЭ.
- Увеличение С-реактивного белка и серомукоида.
- Повышение титров антистрептолизина О (от 1:500 до 1:1000).
- На ЭХО-КГ — увеличение размеров сердца, изменения клапанов.
Лечение хореи Гентингтона
Лечение при данном заболевании носит поддерживающий характер. Препаратами выбора при гиперкинезах являются нейролептики, которые блокируют дофаминовые рецепторы нейронов. Их применение позволяет частично подавить хорею и возбуждение (ажитацию) у больного.

Для лечения применяются типичные нейролептики Галоперидол, Пимозид, Модитен. Менее эффективными являются Сульпирид и Тиаприд, но они имеют меньше побочных действий, поэтому используются в виде препаратов выбора. Также широко применяются атипичные нейролептики — Рисперидон, Клозапин, Оланзапин, Арипипразол, Нормокинезтин. Нейролептики используются в низких дозах, а продолжительночть их применения ограничивается двумя неделями с постепенным уменьшением дозы и отменой.
Применение Нормокинезтина показано при хорее и отсутствии депрессии, агрессивного поведения или психоза. На поздних стадиях, когда развивается мышечная ригидность, препарат не показан, поскольку усиливает этот симптом. Арипипразол (нейролептик третьего поколения) обладает эффективностью сопоставимой с Нормокинезтином, но при этом в меньшей степени вызывает депрессию и седативный эффект.
При умеренном гиперкинезе нейролептики могут не назначаться, а больному рекомендуются препараты, блокирующие глутаматергические рецепторы (Амантадин, Мидантан, Мемантин Канон), антиконвульсанты (Топамакс, Топирамат) и симпатолитики (Резерпин). Также возможна комбинация нейролептик + антиглутаматергический препарат или антиконвульсант + симпатолитик. Существуют данные в пользу того, что длительный прием Мемантина и коэнзима Q10 несколько замедляет прогрессирование заболевания. Важно с помощью нейролептиков и антидепрессантов корригировать сопутствующие психические расстройства, прежде всего депрессию и вспышки агрессии.
При ревматической хорее комплексное лечение включает:
- Антибактериальную терапию (Пенициллин с переводом на формы длительного действия — Экстенциллин, Ретарпен, Бициллин-1, Бициллин-5).
- Противовоспалительную (Преднизолон до 10 дней с постепенным снижением дозы).
- Противоэпилептические препараты (Конвулекс, Депакин, Карбамазепин, Мазепин, Финлепсин). В случае, если они оказываются неэффективными, назначаются нейролептики в минимальной дозе.
- Физиотерапию (электросон, электрофорез с бромидом кальция, УВЧ по лобно-затылочной методике).
Читайте также:
