
Инвалидность при атаксии фридрейха
БОЛЕЗНЬ НАСЛЕДСТВЕННАЯ АТАКСИЯ ФРИДЕРЕЙХА
БОЛЕЗНЬ ФРИДЕРЕЙХА — ОПРЕДЕЛЕНИЕ:
семейная атаксия, — наследственное аутосомно-рецессивное заболевание, при котором в основном поражаются белое вещество и задние корешки спинного мозга, проявляющееся как правило атаксией (т.е. неустойчивостью).
БОЛЕЗНЬ ФРИДЕРЕЙХА — РАСПРОСТРАНЕННОСТЬ:
Относительно редкое заболевание (1 случай на 22 ООО—25 ООО населения). В то же время семейная атаксия фридерейха является лучше всего изученным заболеваниям из группы спино-церебеллярных дегенераций, характеризующихся преобладанием координаторных нарушений, преимущественно спинальной или мозжечковой атаксией. Среди этих заболеваний встречается большое количество переходных форм, в этой связи диагностика нередко весьма затруднена. Социальное значение определяется выраженным нарушением жизнедеятельности и тяжестью инвалидности фактически во всех случаях клинически очерченной болезнью фридерейха с ранним началом.
БОЛЕЗНЬ ФРИДЕРЕЙХА — ПРОИСХОЖДЕНИЕ:
Ген болезни фридерейха расположен на 9-й хромосоме (9ql3 — q21), хотя где именно не найден. Отмечают неполную частоту проявления гена, частоту единичность случаев и изменчивость симптомов в клинической картине заболевания. Первичный биохимический дефект пока неизвестен, но имеются сведения о дефиците витамина Е в сыворотке крови до уровня меньше 1 мкг/мл и нарушении в местоположении определённого гена, кодирующего информацию о а-токоферолтрансферазе, встраивающей а-токоферол в липопротеины (Swanson, 1995). Наряду с поражением белого вещества спинного мозга и корешков спинного мозга в процесс вовлекаются задние и нижние проводники идущие от ядер расположенных в спинном мозге к мозжечку. Обнаруживаются патология мозжечка, дегенерация пирамидных путей, изменения в коре больших полушарий.
БОЛЕЗНЬ ФРИДЕРЕЙХА — КЛИНИЧЕСКАЯ КАРТИНА:
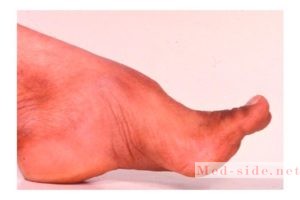
Болезнь фридерейха семейная атаксия начинается обычно в 6—12 летнем возрасте, в редких случаях около 20 (поздние формы, по С. Н. Давиденкову). Первые симптомы — неустойчивость при ходьбе и частые падения. Рано выявляется смешанная сенситивно-мозжечковая атаксия, в дальнейшем— нарушение координации в руках, нарушения речи. Характерно снижение рефлексов с сухожилий вначале ног, затем рук, могут быть умеренно выраженные слабость в конечностях и повышение мышечного тонуса в них. У части пациентов снижается интеллектуальные функции. При исследовании глазного дна окулистом — атрофия зрительных нервов. Характерны утрата суставного — мышечного чувства, нарушение вибрационной чувствительности, такие изменения как — полая стопа, деформации позвоночника, кардиомиопатия.
Диагностические критерии: аутосомно-рецессивное наследование; неустойчивость ходьбы (атаксия) прогрессирующего характера с вовлечением рук; нечеткость при произношении слов; расстройство суставно-мышечного чувства и вибрационного; отсутствие сухожильных рефлексов на ногах. Менее значимые симптомы: полая стопа, сколиоз, кифосколиоз, кардиомиопатия, рефлекс Бабинского — то есть пальцы идут кверху при штрихавом раздражении подошвы стопы.
БОЛЕЗНЬ ФРИДЕРЕЙХА — ДАННЫЕ ДОПОЛНИТЕЛЬНЫХ МЕТОДОВ ОБСЛЕДОВАНИЯ :
— консультации специалистов: терапевта, кардиолога, эндокринолога окулиста, лор врача.
— ЭМГ, ЭНМГ — значительное снижение проведения возбуждения по большеберцовому нерву, уменьшение амплитуды потенциалов мышц и нервов нижних конечностей. Можно выявить различия в скорости проведения импульсов при спиноцеребеллярной атаксии (в случае Болезни Фридерейха) и мозжечковой атаксии Мари. Характерно снижение сомато-сенсорных вызванных потенциалов;
— ЭКГ, эхо-кардиограмма (полная или частичная блокада, порок сердца);
— ЭЭГ — диффузно-пароксизмальное изменение биоэлектрической активности, на этом фоне возможны эпилептические припадки;
— рентгенография скелета;
— МРТ — можно выявить признаки атрофии спинного мозга;
— экспериментально-психологическое исследование — можно выявить изменение интелектуальных функций;
БОЛЕЗНЬ ФРИДЕРЕЙХА — ОТЛИЧИЕ ОТ СХОЖИХ ПО КЛИНИЧЕСКОЙ КАРТИНЕ ЗАБОЛЕВАНИЙ:
1. С фуникулярным миелозом — одним из клинических синдромов гиповитаминоза B12. Имеют значение: пернициозная анемия, заболевания желудочно-кишечного тракта, типичная клиническая картина — комбинированное поражение пирамидных трактов и задних канатиков спинного мозга. Выраженность парезов обычно значительная, отсутствуют типичные для Болезни Фридерейха экстраневральные расстройства. Снижено количество витамина B12 в сыворотке крови. Эффективна терапия цианокобаламином в больших дозах (1000 мкг ежедневно).Со спинной сухоткой ;
2. Со спинной сухоткой ;
3. С синдромом Русси-Леви. В отличие от Болезни Фридерейха доминантный тип наследования, начало на первом году жизни, медленное прогрессирование со стабилизацией процесса в возрасте около 20 лет. В остальном клиническая картина весьма сходна.
4. С группой мозжечковых атаксий:
а) оливопонтоцеребеллярной атрофией (формой Менцеля). В отличие от Болезни Фридерейха наследуется аутосомно-доминантно, начинается после 30 лет; помимо атаксии типичны гиперкинезы, акинетико-ригидный синдром;
б) наследственной мозжечковой атаксией Мари. В настоящее время считается не самостоятельной нозологической формой, а одним из синдромов наследственных мозжечковых атрофии с аутосомно-доминантным типом наследования. В отличие от Болезни Фридерейха наряду с атаксией наблюдается отчетливый интенционный тремор, сохраняются сухожильные рефлексы, нет скелетных аномалий;
в) атаксией-телеангиоэктазией Луи-Бар. Наследование аутосомно-рецессивное. Проявляется в раннем детстве, иногда в 3—6 лет, мозжечковой атаксией. В дальнейшем вследствие распространения процесса на спинной мозг, базальные ганглии наряду с прогрессирующей мозжечковой дискоординацией возникают нарушения глубокой чувствительности, мышечные атрофии, хореоатетоз. Основным дифференциально-диагностическим признаком является стойкое расширение мелких сосудов кожи (артериол, венул, капилляров) невоспалительной природы, проявляющееся сосудистыми звёздочками или сеточками. Диаметр расширенных сосудов составляет 0,5—1 мм. Распологаются они на конъюктиве, лице, руках и ногах. Больные редко доживают до 30—40 лет, инвалидизируются в детском возрасте;
г) ненаследственными мозжечковыми синдромами. Обычно вторичные (алкогольные, паранеопластические, гипотиреоидные). К ним, повидимому, относится и давно описанная поздняя кортикальная мозжечковая атрофия Мари-Фуа-Алажуанина. От Болезни Фридерейха отличаются поздним началом (около 50 лет), признаками полиневропатии, отсутствием изменений скелета. Компьютерная томография выявляет атрофические процессы мозжечка.
Принципы лечения
Показания для госпитализации: необходимость уточнения диагноза, проведение курса поддерживающей терапии в случае декомпенсации. Рекомендуются ноотропы, церебролизин, витаминотерапия. Обнаружено, что назначение а-токоферола в дозе 800 мг в день восстанавливает содержание витамина Е в сыворотке крови до нормальных значений. Важное место занимают лечебная физкультура, особенно корригирующая гимнастика. При выраженной деформации стопы в необходимых случаях ортопедические операции.
БОЛЕЗНЬ ФРИДЕРЕЙХА — ЛЕЧЕНИЕ:
Показания для госпитализации: необходимость уточнения диагноза, проведение курса поддерживающей терапии в случае декомпенсации. Рекомендуются ноотропы, церебролизин, витаминотерапия. Обнаружено, что назначение а-токоферола в дозе 800 мг в день восстанавливает содержание витамина Е в сыворотке крови до нормальных значений. Важное место занимают лечебная физкультура, особенно корригирующая гимнастика. При выраженной деформации стопы в необходимых случаях ортопедические операции.
БОЛЕЗНЬ ФРИДЕРЕЙХА — БОЛЬНИЧНЫЙ ЛИСТ И СРОКИ ПРЕБЫВАНИЯ:
1. Стационарное обследование для уточнения диагноза или степени ограничения жизнедеятельности при решении экспертных вопросов (по направлению МСЭ). больничный лист выдается на 2—3 недели.
2. Декомпенсация, вызванная различными причинами, у работающих больных (больничный лист выдается на — 3—4 недели).
3. В случае ортопедической операции (больничный лист выдается в этом случае в зависимости от характера оперативного вмешательства).
БОЛЕЗНЬ ФРИДЕРЕЙХА — ПРОТИВОПОКАЗАННЫЕ РАБОТЫ И УСЛОВИЯ ТРУДА:
Работы, требующие тонких, точных движений (гравер, машинистка, сборщик радиосхем и т. п.), у конвейера, водительские и другие, связанные с ответственностью за безопасность движения,требующие длительного речевого контакта, многие виды интеллектуального труда.
БОЛЕЗНЬ ФРИДЕРЕЙХА — ПОКАЗАНИЯ ДЛЯ ОФОРМЛЕНИЕ ИНВАЛИДНОСТИ ВО МСЭ:
1. У ребенка — социальная недостаточность, требующая социальной защиты и помощи при диагностике болезни семейной атаксии Фридерейха.
2. У взрослых больных — явное нарушение жизнедеятельности или ее ограничение, в основном по причине неустойчивости.
БОЛЕЗНЬ ФРИДЕРЕЙХА — МИНИМУМ ДОПОЛНИТЕЛЬНЫХ ОБСЛЕДОВАНИЙ ПРИ НАПРАВЛЕНИИ НА МЕДИКО — СОЦИАЛЬНУЮ ЭКСПЕРТИЗУ ДЛЯ ОФОРМЛЕНИИ ГРУППЫ ИНВАЛДНОСТИ (ДЛЯ МСЭ):
1. Рентгенография скелета.
2. Данные осмотра окулиста, отоларинголога.
3. ЭКГ, Эхо-КГ.
4. ЭНМГ, ЭМГ.
5. Консультация терапевта (кардиолога), при необходимости эндокринолога, психиатра.
6. Экспериментально-психологическое исследование (по показаниям).
БОЛЕЗНЬ ФРИДЕРЕЙХА КРИТЕРИИ УСТАНОВЛЕНИЯ ГРУППЫ ИНВАЛИДНОСТИ:
БОЛЕЗНЬ ФРИДЕРЕЙХА — ПРОФИЛАКТИКА ИНВАЛИДНОСТИ:
1. Первичная профилактика: а) медико-генетическое консультирование (учитывается возможность рождения больного ребенка у здоровых родителей в связи с аутосомно-рецессивным типом наследования); б) пренатальная диагностика.
2. Вторичная профилактика: а) своевременная диагностика, диспансерное наблюдение и регулярная поддерживающая терапия; б) при стертых, абортивных, иногда поздних формах БФ возможна профориентация (продавец, кладовщик, администратор, контролер и т. п.).
3. Третичная профилактика: а) предупреждение срывов компенсации у работающих больных; б) своевременное определение инвалидности с учетом степени ограничения жизнедеятельности; в) осуществление других мер социальной защиты.
БОЛЕЗНЬ ФРИДЕРЕЙХА — РЕАБИЛИТАЦИЯ:
1. Медицинская реабилитация: а) регулярная симптоматическая медикаментозная терапия, лечебная физическая культура; б) при необходимости хирургическое лечение и снабжение ортопедической обувью.
2. Профессиональная реабилитация: инвалиды третьей группы при потере профессии могут выполнять нетяжелые, подсобные (уборка небольшого помещения), несложные ремонтные и слесарные работы и т. п.
3. Социальная реабилитация: Индивидуальная программа реабилитации с обеспечением средствами передвижения (креслом-коляской, креслом-каталкой), обучение самообслуживанию; психологическая помощь семье, обучение уходу за тяжелым больным.

| Мои статьи [3] |
| Медико-социальная экспертиза при некоторых заболеваниях [170] |
Войти через uID
Определение
Болезнь Фридрейха (БФ), семейная атаксия, — наследственное аутосомно-рецессивное заболевание, обусловленное преимущественной дегенерацией задних столбов и задних корешков спинного мозга, проявляющееся в основном атаксией.
Относится к сравнительно редким (1 : 22000—25000 населения), но лучше всего изученным заболеваниям из группы спино- церебеллярных дегенераций, характеризующихся преобладанием координаторных нарушений, преимущественно спинальной или мозжечковой атаксией. Среди них имеется много переходных форм, в связи с чем клиническая диагностика нередко весьма затруднена. Социальное значение определяется выраженным нарушением жизнедеятельности и тяжестью инвалидности фактически во всех случаях клинически очерченной БФ с ранним началом.
Этиология, патоморфология
Ген БФ картирован на 9-й хромосоме (9ql3 — q21), но не идентифицирован. Отмечают неполную пенетрантность, частоту спорадических случаев и вариабельность клинической картины заболевания. Первичный биохимический дефект пока неизвестен, однако есть данные о снижении концентрации витамина Е в сыворотке крови до уровня менее 1 мкг/мл и нарушении в локусе, кодирующем информацию о а-токоферолтрансферазе, встраивающей а-токоферол в липопротеины (Swanson, 1995). Наряду с поражением задних канатиков и корешков в процесс вовлекаются дорсальные и вентральные спиноцеребеллярные пути. Обнаруживаются патология мозжечка, дегенерация пирамидных путей, изменения в коре больших полушарий.
Клиника и критерии диагностики
Начало БФ обычно в 6—12 лет, реже около 20 (поздние формы, по С. Н. Давиденкову). Первые симптомы — неустойчивость при ходьбе, частые падения. Рано выявляется смешанная сенситивно-мозжечковая атаксия, в дальнейшем—дискоординация в руках, нарушения речи. Характерна гипорефлексия вначале на нижних конечностях, затем на верхних, могут быть умеренно выраженные центральные парезы. У части больных снижается интеллект, выявляется атрофия зрительных нервов.
Типичны утрата мышечно-суставного чувства, нарушение вибрационной чувствительности, экстраневральные симптомы (полая стопа, деформации позвоночника, кардиомиопатия).
Диагностические критерии: аутосомно-рецессивное наследование; атаксия ходьбы прогрессирующего характера с вовлечением верхних конечностей; дизартрия; расстройство мышечно-суставного чувства и вибрации; выпадение глубоких рефлексов на ногах.
Менее значимые симптомы: полая стопа, сколиоз, кифосколиоз, кардиомиопатия, рефлекс Бабинского.
Данные дополнительных исследований:
— рентгенография скелета;
— ЭМГ, ЭНМГ — значительное снижение проведения возбуждения по большеберцовому нерву, уменьшение амплитуды потенциалов мышц и нервов нижних конечностей. Можно выявить различия в скорости проведения импульсов при спиноцеребеллярной атаксии (в случае БФ) и мозжечковой атаксии Мари. Характерно снижение сомато-сенсорных вызванных потенциалов;
— ЭКГ, эхо-кардиограмма (полная или частичная блокада, порок сердца);
— ЭЭГ — диффузно-пароксизмальное изменение биоэлектрической активности, на этом фоне возможны эпилептические припадки;
— МРТ — атрофия спинного мозга;
— экспериментально-психологическое исследование (выявление когнитивного дефекта);
— консультация терапевта, кардиолога (диагностика кардио- миопатии), эндокринолога;
— обследование у окулиста, отоларинголога.
Дифференциальный диагноз
1.Со спинной сухоткой;
2.С фуникулярным миелозом — одним из клинических синдромов гиповитаминоза В12. Имеют значение: пернициозная анемия, заболевания желудочно-кишечного тракта, типичная клиническая картина — комбинированное поражение пирамидных трактов и задних канатиков спинного мозга. Выраженность парезов обычно значительная, отсутствуют типичные для БФ экстраневральные расстройства. Снижено содержание цианокобаламина (витамина B12) в сыворотке крови. Эффективна терапия цианокобаламином в больших дозах (1000 мкг ежедневно).
3.С синдромом Русси-Леви. В отличие от БФ доминантный тип наследования, начало на первом году жизни, медленное прогрессирование со стабилизацией процесса в возрасте около 20 лет. В остальном клиническая картина весьма сходна.
4.С группой мозжечковых атаксий:
а) оливопонтоцеребеллярной атрофией (формой Менцеля). В отличие от БФ наследуется аутосомно-доминантно, начинается после 30 лет; помимо атаксии типичны гиперкинезы, акинетико- ригидный синдром;
б) наследственной мозжечковой атаксией Мари. В настоящее время считается не самостоятельной нозологической формой, а одним из синдромов наследственных мозжечковых атрофий с аутосомно-доминантным типом наследования. В отличие от БФ наряду с атаксией наблюдается отчетливый интенционный тремор, сохраняются сухожильные рефлексы, нет скелетных аномалий;
в) атаксией-телеангиоэктазией Луи-Бар. Наследование аутосомно-рецессивное. Проявляется в раннем детстве, иногда в 3—6 лет, мозжечковой атаксией. В дальнейшем вследствие распространения процесса на спинной мозг, базальные ганглии наряду с прогрессирующей мозжечковой инкоординацией возникают нарушения глубокой чувствительности, мышечные атрофии, хореоатетоз. Основным дифференциально-диагностическим признаком являются телеангиоэктазии (на конъюктиве, коже лица, конечностей). Больные редко доживают до 30—40 лет, инвалидизируются в детском возрасте;
г) ненаследственными мозжечковыми синдромами. Обычно вторичные (алкогольные, паранеопластические, гипотиреоидные). К ним, по-видимому, относится и давно описанная поздняя кортикальная мозжечковая атрофия Мари-Фуа-Алажуанина. От БФ отличаются поздним началом (около 50 лет), признаками полиневропатии, отсутствием изменений скелета. На КТ выявляется атрофия мозжечка.
Течение и прогноз
БФ может прогрессировать очень быстро (особенно при раннем начале), приводя через 5—6 лет к полной обездвиженности и зависимости от окружающих. В других случаях заболевание развивается медленно и больные даже через 15 лет от начала болезни сохраняют способность самостоятельного передвижения, несмотря на выраженное нарушение жизнедеятельности. По нашим данным, инвалидность II и I группы иногда определяется лишь через 20 лет. Прогноз при развернутой клинической картине БФ неблагоприятен. Больные погибают от интеркуррентных заболеваний, кардиомиопатии.
Принципы лечения
Показания для госпитализации: необходимость уточнения диагноза, проведение курса поддерживающей терапии в случае декомпенсации. Рекомендуются ноотропы, церебролизин, витаминотерапия. Обнаружено, что назначение сс-токоферола в дозе 800 мг в день восстанавливает содержание витамина Е в сыворотке крови до нормальных значений. Важное место занимают лечебная физкультура, особенно корригирующая гимнастика. При выраженной деформации стопы в необходимых случаях ортопедические операции.
Медико-социальная экспертиза Критерии ВУТ
1.Стационарное обследование для уточнения диагноза или степени ограничения жизнедеятельности при решении экспертных вопросов (по направлению БМСЭ). ВН в течение 2—3 недель.
2.Декомпенсация, вызванная различными причинами, у работающих больных (ВН — 3—4 недели).
3.В случае ортопедической операции (продолжительность ВН зависит от характера оперативного вмешательства).
Характеристика ограничения жизнедеятельности
Основным критерием оценки жизнедеятельности является смешанная (сенситивно-мозжечковая) атаксия.
Целесообразно выделять 4 степени ее выраженности:
легкая — координаторные расстройства выявляются лишь при выполнении специальных заданий, тестов;
умеренная — инкоординация проявляется при выполнении обычных движений;
выраженная — значительно затрудняется выполнение обычных движений;
резко выраженная — невозможно или нерезультативно выполнение обычных движений.
В случае преобладания сенситивного компонента атаксии дополнительно учитывается степень выпадения мышечно-суставного чувства:
легкие нарушения — больной ошибается в определении движения пальца при пороговой амплитуде (2 градуса);
умеренные — не определяет пороговых движений, для правильного ответа нужны движения под углом больше 2 градусов;
выраженные — не определяет движения в пальцах стоп и в голеностопных суставах;
резко выраженные — не определяет движения в коленных и тазобедренных суставах.
В зависимости от выраженности атаксия приводит к нарушению способности к передвижению, пользованию транспортом, выполнению точных координированных движений верхними конечностями, что ведет к снижению ручной активности. Поэтому рано нарушается возможность трудовой деятельности, а затем снижается и утрачивается способность к личному уходу.
Нарушение речевой функции, интеллектуальный дефект, прогрессирующие с течением болезни, ограничивают возможность общения, трудовой деятельности, а также способность к обучению, что затрудняет профессиональную реабилитацию. К нарушению самообслуживания различной степени может приводить и кардиальная патология.
В меньшей степени жизнедеятельность ограничивают другие симптомы БФ: костные аномалии, зрительные, слуховые расстройства, парезы, тазовые нарушения. Однако и они должны быть учтены при решении вопроса об инвалидности наряду с особенностями течения болезни (темпом прогрессирования), социальными факторами (наличие профессии). Встречаются больные с абортивными симптомами поражения нервной системы или лишь с более или менее выраженными костными аномалиями, когда жизнедеятельность сохранена, трудоспособность существенно не ограничена.
Противопоказанные виды и условия труда
Работы, требующие тонких, точных движений (гравер, машинистка, сборщик радиосхем и т. п.), у конвейера, водительские и другие, связанные с ответственностью за безопасность движения, требующие длительного речевого контакта, многие виды интеллектуального труда.
Показания для направления на БМСЭ
1.Социальная недостаточность, требующая социальной защиты и помощи при диагностике БФ у ребенка.
2.У взрослых больных — выраженное нарушение жизнедеятельности или ее ограничение, главным образом вследствие атаксии.
Необходимый минимум обследования при направлении на БМСЭ
1.Рентгенография скелета.
2.Данные осмотра окулиста, отоларинголога.
3.ЭКГ, Эхо-КГ.
4.ЭНМГ, ЭМГ.
5.Консультация терапевта (кардиолога), при необходимости эндокринолога, психиатра.
6.Экспериментально-психологическое исследование (по показаниям).
КРИТЕРИИ ИНВАЛИДНОСТИ ПРИ БОЛЕЗНИ ФРИДРЕЙХА У ВЗРОСЛЫХ В 2020 ГОДУ
Инвалидность не устанавливается в случае, если у больного имеются:
Незначительные нарушения координации и равновесия, целевой моторики; незначительные легкие сенситивно-мозжечковые нарушения, выявляемые только при выполнении специальных тестов, не нарушающие адаптацию и функционирование человека.
Незначительные нарушения функций организма.
Инвалидность 3-й группы устанавливается в случае, если у больного имеются:
Умеренные нарушения координации и равновесия, целевой моторики; умеренные сенситивно-мозжечковые нарушения, выявляемые при выполнении обычных движений, нарушающие опору и передвижение.
Умеренные нарушения функций организма.
Инвалидность 2-й группы устанавливается в случае, если у больного имеются:
Выраженные нарушения координации и равновесия, целевой моторики; выраженные сенситивно-мозжечковые нарушения (значительно затрудняется передвижение, выполнение обычных движений).
Выраженные нарушения функций организма.
Инвалидность 1-й группы устанавливается в случае, если у больного имеются:
Значительно выраженные нарушения координации и равновесия, целевой моторики; значительно выраженные сенситивно-атактические нарушения.
Значительно выраженные нарушения функций организма.
КРИТЕРИИ ИНВАЛИДНОСТИ ПРИ БОЛЕЗНИ ФРИДРЕЙХА У ДЕТЕЙ В 2020 ГОДУ
Инвалидность не устанавливается в случае, если у ребенка имеются:
незначительные нарушения координации и равновесия, целевой моторики; незначительные легкие сенситивно-мозжечковые нарушения, выявляемые только при выполнении специальных тестов, не нарушающие адаптацию и функционирование ребенка.
Незначительные нарушения функций организма.
Категория "ребенок-инвалид" устанавливается в случае, если у больного имеются:
- умеренные нарушения координации и равновесия, целевой моторики; умеренные сенситивно-мозжечковые нарушения, выявляемые при выполнении обычных движений, нарушающие опору и передвижение, препятствующие формированию возрастных навыков.
Умеренные нарушения функций организма.
- выраженные нарушения координации и равновесия, целевой моторики; выраженные сенситивно-мозжечковые нарушения (значительно затрудняется передвижение, выполнение обычных движений).
Выраженные нарушения функций организма.
- значительно выраженные нарушения координации и равновесия, целевой моторики; значительно выраженные сенситивно-атактические нарушения.
Значительно выраженные нарушения функций организма.
Получить официальное заключение о наличии (или отсутствии) оснований для установления инвалидности больной может только по результатам своего освидетельствования в бюро МСЭ соответствующего региона.
Порядок оформления документов для прохождения МСЭ (включая и алгоритм действий при отказе лечащих врачей направлять больного на МСЭ) достаточно подробно расписан в этом разделе форума: Оформление инвалидности простым языком
Атаксия Фридрейха – это наследственное заболевание, которое встречается в неврологической практике крайне редко, менее, чем в 1% случаев. Для того, чтобы оно проявилось, необходимо наличие рецессивного гена у обоих родителей ребенка. Патология впервые дает о себе знать в 10-13-летнем возрасте.
Лечение атаксии Фридрейха – симптоматическое. Ребенок рано становится инвалидом, а средняя продолжительность жизни составляет до 15 лет с момента начала заболевания. Но были зафиксированы случаи, в которых пациенты доживали до 70-80 лет.
Опасность заключается в том, что болеет данной атаксией 1 на 100 тысяч населения, а мутация есть у 1 из 100 человек.
Причины
Тип наследования данного заболевания – аутосомно-рецессивный. Это значит, что оба родителя ребенка должны иметь рецессивный ген, но сами могут не иметь атаксию Фридрейха. Мужчины и женщины болеют одинаково часто.
Происходит мутация в 9 хромосоме. Затем нарушаются процессы оксигенации фермента ЛДГ, снижается синтез медиатора нервной системы – ацетилхолина в спинном мозге, столбе, мозжечке. Нарушается процесс транспорта железа в митохондрии из-за недостаточности белка фратаксина. Происходит снижение глютамина на определенных участках головного мозга.
Патанатомически происходит дегенерация спинного мозга, повреждаются боковые канатики. Это – результат действия свободных радикалов. Свободные радикалы также поражают клетки сердца, поджелудочной железы. Антиоксидантные системы организма не успевают обезвредить радикалы.
Симптомы
Первый симптом патологии – неустойчивость походки. Спиноцеребеллярная наследственная или семейная атаксия Фридрейха развивается очень медленно. Пациенту на 4-5 год проявления болезни понадобится инвалидное кресло.
В неврологии выделяют основные симптомы атаксии Фридрейха:
- сенситивно-мозжечковая атаксия,
- нистагм,
- гипотония мышц,
- арефлексия.
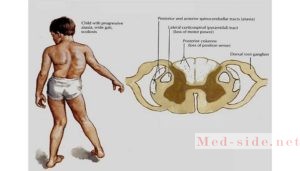
После манифестации болезни у пациента выпадает вибрационная и глубокая мышечно-суставная чувствительность. Затем расстройства координации становятся более заметными. Шаткость при ходьбе сопровождается спотыканиями и падениями. Появляется дрожь, тремор в руках, что сопровождается искривлением почерка.
Затем присоединяются нарушения речевого аппарата и слуха (нейросенсорная тугоухость).
Деформируются пальцы рук и ног, развивается косолапость.
Диагностика
У пациента понижен мышечный тонус, наблюдается снижение сухожильных рефлексов. Исчезает коленный рефлекс, что является ранним признаком атаксии Фридрейха.
Проверить сухожильные рефлексы дома можно самостоятельно. Достаточно ребром ладони легко ударить по сухожилью под коленной чашечкой (на 1,5-2 см ниже). Сначала их проверяют на 1 потом на 2 ноге. При этом пациента просят закинуть ногу на ногу поочередно и расслабиться.
Проверяют устойчивость в позе Ромберга, пациент с атаксией , неустойчив. Пальце-носовая проба также отрицательная, пациент промахивается с закрытыми глазами.
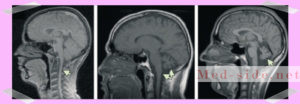
После первичной диагностики у невролога, пациент попадает на МРТ. На снимках визуализируются атрофические процессы в продолговатом мозге и мосте, атрофированный мозжечок. То есть, его размеры уменьшены. Также уменьшается поперечник спинного мозга.
На электромиографии отмечается снижение проводимости по чувствительным волокнам.
Для исследования состояния 9 хромосомы назначается кариотипирование и ПЦР. Дополнительно назначаются консультации у офтальмолога, эндокринолога, кардиолога, делается УЗИ сердца или ЭхоКГ.
Можно определить вероятность атаксии Фридрейха у плода. Проводится ДНК-диагностика ворсин хориона на 8-12 неделе или же для анализа необходим образец амниотической жидкости на 16-24 неделе беременности.
Дифференциальный диагноз
Дифференциальную диагностику проводят с наследственными заболеваниями мозжечка. А именно с такими атаксиями, как:
- наследственная атаксия Пьера-Мари,
- клинические формы оливо-понто-церебеллярной дегенерации,
- болезнь Рефсума,
- болезнь Руси-Левина,
- болезнь Маринеску-Шагрена.
Дополнительно исключают такие патологии, как: опухоль мозжечка, фуникулярный миелоз из-за недостаточности витамина В12, рассеянный склероз, синдром Луи-Барр (атаксия-телеангиоэктазии), сифилис ЦНС.
Клинические рекомендации
Назначают препараты, которые улучшают метаболизм миокарда или сердечной мышцы , тиаминпирофосфат, инозин, триметазидин, 5-гидроксипрофан.
Для улучшения кровообращения в сосудах головного мозга назначают ноотропы и нейропротекторы: энцефабол, гамма-аминомасляную кислоту, пирацетам, меклофеноксат, пиритинол, поливитамины. Уделяют внимание ЛФК и массажам. Иногда проводят хирургическую коррекцию нарушений опорно-двигательного аппарата.
Пиоглитазон и Идебенон – препараты, которые владеют антиоксидантной активностью и содержат коэнзим Q вместе с витамином E. К сожалению, клиническая эффективность этих средств не подтверждена. Об исследованиях можно прочесть здесь.
Поэтому лечение атаксии Фридрейха остается симптоматическим, направленным на поддержание жизни пациента. В случае аритмий необходимо назначить блокаторы кальциевых каналов, антиаритмики. При сахарном диабете – диету с возможным последующим подключением инсулина.
Разрабатывается в Висконсине новая методика лечения, которая предлагает встраивать в ДНК синтетический фактор элонгации. Эксперименты на клетках пациентов с атаксией Фридрейха показали, что Syn-TEF1 , фактор элонгации способен полностью восстановить экспрессию гена FXN.
Патология является неизлечимой. Можно замедлить течение болезни, но в среднем прогноз для жизни составляет 10-15 лет с момента манифестации, то есть клинического проявления атаксии Фридрейха.
Только при отсутствии сахарного диабета и других патологий, особенно со стороны сердечно-сосудистой системы, пациент может дожить до 70-80 лет. Продолжительность жизни с атаксией Фридрейха у женщин благоприятнее, они живут на 30-40% дольше, чем мужчины.
Читайте также:
