
Невропатия с параличами от сдавления
Автор - д.м.н. Елена Леонидовна Дадали Письмо Автору
НАСЛЕДСТВЕННАЯ НЕЙРОПАТИЯ СО СКЛОННОСТЬЮ К ПАРАЛИЧАМ ОТ СДАВЛЕНИЯ (OMIM: 162500)
Впервые описал De Jong в 1947 году у членов трех поколений семьи. Заболевание является аллельным вариантом НМСН 1А типа, так как обусловлено мутациями, захватывающими область локализации гена РМР 22.
Заболевание возникает в возрасте от 8 до 64 лет (средний возраст - 26 лет) и характеризуется рецидивирующими парезами периферических нервов, возникающими остро после небольших травм или сдавлений. У 40% больных парезы возникают после интенсивных физических нагрузок, а у 10 % при пробуждении. Продолжительность двигательных нарушений колеблется от одного дня до нескольких месяцев. У 50 % больных в патологический процесс вовлекаются два и более периферических нерва, патологический процесс часто бывает асимметричным. В редких случаях в процесс вовлекаются черепно-мозговые нервы, особенно бульбарная группа, что проявляется парезом голосовых связок или лицевой мускулатуры. У 62% больных сухожильные рефлексы с рук и ног не изменяются, у 30-35% больных выявляется их умеренное снижение, и лишь у 12% больных сухожильные рефлексы исчезают. Характерно возникновение расстройств чувствительности, особенно выраженное в период существования пареза.
Описаны больные с наличием сколиоза и деформации стоп по типу полых, а также с глухотой.
На электромиограмме выявляется умеренное увеличение латенции по срединному нерву, снижение скоростей проведения по сенсорным и моторным нервам.
При морфологическом изучении биоптата периферических нервов выявляются изменения сходные с таковыми при наследственной моторно- сенсорной нейропатии 1 типа. Характерно возникновение очагов демиелинизации и образование томакул - колбасовидных утолщений миелиновой оболочки периферических нервов. Их наличие отмечено как в моторных, так и сенсорных нервах. Считается, что томакулы формируются в период раннего развития ребенка и отражают нарушение взаимодействия аксона и миелина.
Тип наследования аутосомно-доминантный.
Заболевание является аллельным вариантом наследственной моторно - сенсорной нейропатией 1 А типа, так как обусловлено делецией в области хромосомы 17 р11.2-12, захватывающей область локализации гена РМР22 или точковыми мутациями в этом гене. Делеции протяженностью 1,5 кб встречаются у 85 % больных. 37% случаев - спорадические. Характерна неполная пенетрантность и варьирующая экспрессивность.
Возможна дородовая диагностика с использованием молекулярно- генетических методов.
- Chance, P. F.; Alderson, M. K.; Leppig, K. A.; Lensch, M. W.; Matsunami, N.; Smith, B.; Swanson, P. D.; Odelberg, S. J.; Disteche, C. M.; Bird, T. D.: DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 72: 143-151, 1993.
- De Jong, J. G. Y.: Over families met hereditarie disposite tot het optreten van neuritiden, gecorreleard met migraine. Psychiat. Neurol. Bl. 50: 60-76, 1947.
- Mariman, E. C. M.; Gabreels-Festen, A. A. W. M.; van Beersum, S. E. C.; Jon- gen, P. J. H.; Ropers, H.-H.; Gabreels, F. J. M.: Gene for hereditary neuropathy with liability to pressure palsies (HNPP) maps to chromosome 17 at or close to the locus for HMSN type 1. Hum. Genet. 92: 87-90, 1993.
- Mariman, E. C. M.; Gabreels-Festen, A. A. W. M.; van Beersum, S. E. C.; Valentijn, L. J.; Baas, F.; Bolhuis, P. A.; Jongen, P. J. H.; Ropers, H. H.; Gabreels, F. J. M.: Prevalence of the 1.5-Mb 17p deletion in families with hereditary neu- ropathy with liability to pressure palsies. Ann. Neurol. 36: 650-655, 1994.
- Umehara, F.; Kiwaki, T.; Yoshikawa, H.; Nishimura, T.; Nakagawa, M.; Ma- tsumoto, W.; Hashimoto, K.; Izumo, S.; Arimura, Y.; Arimura, K.; Kuriyama, M.; Osame, M.: Deletion in chromosome 17p11.2 including the peripheral myelin protein-22 (PMP-22) gene in hereditary neuropathy with liability to pressure pal- sies. J. Neurol. Sci. 133: 173-176, 1995.
- Verhalle, D.; Lofgren, A.; Nelis, E.; Dehaene, I.; Theys, P.; Lammens, M.; Dom, R.; Van Broeckhoven, C.; Robberecht, W.: Deletion in the CMT1A locus on chromosome 17p11.2 in hereditary neuropathy with liability to pressure palsies. Ann. Neurol. 35: 704-708, 1994.
Что такое "Наследственные полинейропатии"?
Наследственные полинейропатии – это большая группа клинически разнообразных полинейропатий с возможным вовлечением различных органов и систем, в том числе и центральной нервной системы, которые развиваются в результате мутаций (поломок) в генах человека.
Какие мифы существуют среди населения относительно наследственных заболеваний?
- "Наследственные заболевания развиваются с рождения или в раннем детстве"
Это не так - дебют наследственной болезни возможен и во взрослом возрасте, в том числе и после 50 лет. Более того, зачастую пациенты не могут точно назвать возраст начала заболевания, так как симптомы развиваются незаметно. - "Наследственная патология должна быть и у близких родственников. А если в роду ни у кого нет, значит и у меня тоже нет".
Это не так - наследственные заболевания передаются разными путями: по аутосомно-доминантному, аутосомно-рецессивному, Х-сцепленному типу, также возможен митохондриальный тип наследования и др. Часто в семье есть асимптомные носители мутантного гена без клинических признаков заболевания, а симптомы болезни могут быть только у одного из членов семьи. Поэтому отсутствие отягощенного семейного анамнеза не исключает наследственный генез заболевания.

Какие бывают "Наследственная полинейропатии"?
Наследственных полинейропатий очень много, причиной из развития могут быть мутации более чем в 100 генах. Классифицируют наследственные нейропатии в основном по типу наследования, вовлечению тех или иных нервных волокон и характеру их поражения.
Ниже представлена лишь часть из них, преимущественно тех, которые на практике могут диагностироваться в возрасте старше 20 лет:
- Наследственные моторно-сенсорные нейропатии (НМСН)
- Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС)
- Наследственные сенсорные и автономные нейропатии (НСАН)
- Болезнь Фабри
- Транстиретиновая семейная амилоидная нейропатия (ТТР-САП)

Как часто диагностируются наследственные полинейропатии среди населения?
Частота встречаемости всех форм НМСН варьирует от 10 до 40 случаев на 100 000 населения в различных популяциях. На НМСН 1 типа приходится до 70% всех случаев НМСН. Распространенность ННСПС 2-5 случая на 100 000 населения. Болезнь Фабри - 1 случай на 40 000 - 60 000 мужчин. По примерным подсчетам в США встречаемость ТТР-САП составляет 1 случай на 100 000 человек. Следует отметить, что настороженность относительно дебюта наследственной патологии во взрослом возрасте среди врачей разных специальностей достаточно низкая, поэтому некоторые цифры распространенности могут быть занижены.
Какие симптомы наблюдаются у пациентов с наследственными полинейропатиями?
Наследственные заболевания периферических нервов очень разнообразны по клиническим проявлениям. Ниже представлены данные некоторых из них.
Наследственная моторно-сенсорная нейропатия (НМСН) I типа (болезнь Шарко-Мари-Тута) – наиболее часто диагностируемая (в том числе во взрослом возрасте) наследственная полинейропатия, в основе развития которой лежит дефект гена PMP22, в результате чего повреждается миелиновая оболочка периферических нервов. Заболевание чаще всего имеет аутосомно-доминантный тип наследования. Соотношение мужчин и женщин с НМСН 1 типа примерно равно. Симптомы заболевания появляются на первом десятилетии жизни в 75% случаев, в начале второго десятилетия реже - до 10-25%. Обычно чем позднее манифестирует заболевание, тем более благоприятно оно протекает.
Клиническая картина НМСН 1 типа:
- пациентов беспокоят болезненные спазмы мышц голеней (крампи), слабость и деформация стоп, изменение походки, затруднения при беге или подъеме по лестнице;
- постепенно слабость развивается в кистях, в результате чего появляются затруднения при застегивании пуговиц, открывании двери ключом и т.д. В целом руки вовлекаются не ранее чем через 10 лет после появления первых симптомов болезни;
- в меньшей степени беспокоит онемение кистей и стоп, часто эти изменения игнорируются пациентом.
Особенностью является наличие несоответствия жалоб пациента (они минимальны) и неврологического дефицита (он более выражен), т.к. пациент за длительный период прогрессирования заболевания привыкает к тем или иным нарушениям.
При осмотре обращает внимание наличие деформации стоп, контрактур ахилловых сухожилий.
Клиническая картина других типов НМСН и НСАН вариабельна и определяется генетическим дефектом.
Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС) - также часто диагностируемая во взрослом возрасте наследственная нейропатия (дебютирует, как правило, на 2–3-м десятилетии жизни), обусловленная мутацией в гене PMP22, характеризуется повышенной "чувствительностью" периферических нервов к сдавлению в костно-фиброзных каналах (туннелях), что приводит к повторяющимся эпизодам компрессионных туннельных мононевропатий.
Возможно поражение любого периферического нерва, но чаще всего сдавлению подвергаются:
- лучевой нерв на уровне плеча (спиральный канал), при этом развивается слабость разгибателей кисти, кисть "висит" как плеть, возникает онемение наружного края предплечья и кисти;
- малоберцовый нерв на уровне коленного сустава (фибулярный канал), при этом развивается слабость разгибателей стопы, стопа начинает "шлепать", возникает онемение наружного края голени и стопы;
- локтевой нерв на уровне локтевого сустава (кубитальный канал), при этом развивается слабость межкостных мышц кисти, мышцы - отводящих мизинец и других, возникает онемение 4 и 5 пальцев кисти;
- срединный нерв на уровне запястья (карпальный канал), при этом развивается слабость мышц возвышения большого пальца, возникает онемение 1-3 пальцев кисти, характерен болевой синдром;
- и т.д. с развитием соответствующей клинической картины поражения того или иного нерва.
Степень поражения нерва (защемления в канале) может быть различной - от незначительной, когда беспокоит только онемение и парестезии, до более выраженной, когда помимо чувствительных нарушений развивается и слабость мышц.
Развитию симптомов, как правило, предшествует незначительная травма, нахождение длительное время в неудобной статической поза (на четвереньках, на корточках, облокотившись на локоть и др.), ношение неудобной одежды, непривычная физическая нагрузка и прочее.
Мышечная слабость, как правило, сохраняется в течение нескольких дней или недель, а затем сила мышц постепенно восстанавливается (в течение нескольких недель или месяцев). Эпизоды "защемлений" нервов имеют тенденцию к повторению (всего у пациентов может быть от 1 до 10 таких эпизодов). Следует учитывать, что часто лечащие врачи не задумываются о том, что причиной рецидивирующих (повторяющихся) туннельных невропатий – является наследственная патология, что приводит к поздней диагностике и неоправданным медицинским вмешательствам (операциям по поводу туннельных невропатий).
Болезнь Фабри - редкое генетическое заболевание, обусловленное мутацией гена GLA, картированного на длинном плече хромосомы Хq 22.1 и контролирующего структуру фермента альфа-галактозидазы А. Нередко начало заболевания отмечается в подростковом возрасте. Тип наследования - сцепленное с Х-хромосомой.
Клиническая картина:
- жгучая колющая боль в ладонях и стопах; острые приступы мучительной боли в кистях и стопах (кризы Фабри);
- нарушение потоотделения; непереносимость жары/холода;
- сыпь на коже (ангиокератомы);
- помутнение роговицы в виде завитка, которое не ослабляет зрение;
- шум в ушах, потеря слуха;
- желудочно-кишечные расстройства, диарея;
- кардиологические проявления (включая увеличенное сердце и нарушение ритма);
- нарушение функции почек, которое в конечном итоге приводит к терминальной стадии хронической почечной недостаточности;
- нарушение мозгового кровообращения (чаще ишемический инсульт, но может быть и внутримозговое кровоизлияние, или венозный тромбоз).
Транстиретиновая семейная амилоидная полинейропатия (ТТР-САП) – редкое, прогрессирующее, аутосомно-доминантное заболевание, характеризующееся отложением мутантного амилоидного белка транстиретина (TTR) в тканях сердца, кишечника, стекловидного тела и периферических нервов. Заболевание обусловлено точковой мутацией в гене TTR, который кодирует одноименный белок. Симптоматика и степень выраженности нарушений чрезвычайно полиморфны.
Существуют “красные флаги” заболевания: отягощенный семейный анамнез, автономные нарушения (ортостатическая гипотензия, эректильная дисфункция, нарушение потоотделения), нарушение работы органов пищеварительного тракта (запоры, диарея, тошнота) и мочеиспускания, поражение сердца (рестриктивная кардиомиопатия, аритмии, блокады сердца), синдром карпального канала (идиопатический, часто двусторонний), потеря массы тела, помутнение стекловидного тела, прогрессирующая полинейропатия:
-
при раннем дебюте заболевания (
Редкое наследственное заболевание, начинающееся в возрасте старше 10—20 лет и характеризующееся повышенной чувствительностью периферических нервов к сдавлению, что приводит к повторяющимся эпизодам компрессионных невропатий. Чаще сдавлению подвергаются малоберцовый, лучевой, локтевой нервы. Слабость мышц, иннервируемых поврежденным нервом, сохраняется в течение нескольких дней или недель, затем постепенно регрессирует, первоначально с полным восстановлением в результате повторяющихся эпизодов может накапливаться остаточный дефект, и со временем часто развивается клиническая картина, напоминающая болезнь Шарко—Мари—Тута. Патоморфологически выявляются сегментарная демиелинизация и ремиелинизация с локальным утолщением миелиновой оболочки и нарушением ее внутренней структуры. Ген заболевания обнаружен в 17-й хромосоме. Наследование по аутосомно-доминантному типу. Лечение симптоматическое.







О нас
- О компанииРуководство компании
- Молодежь против наркотиков
- Уровень воды в дону
- Грушинский фестиваль
- На самарской набережной
Наши партнеры
- Что и как вам пить
Разделы
- Жизнь
- Красота
- Дети
- ПитаниеКонсультации
- ТестыОбсужденияЗапись к врачу
- Все разделы
ВитаПортал — сайт о здоровье
Мы предоставляем информацию по следующим основным разделам.
- Новости здоровья, питания, диет и здорового образа жизни
- Правильное питание, похудение, диеты
- Аллергия и новые методы лечения
- Вредные привычки и способы отказа от них
- Заболевания человека, методы диагностики и лечения
- Рождение и воспитание детей
- Спорт и фитнес
- Рецепты здорового питания
- Бесплатные консультации врачей
- Блоги врачей, экспертов по питанию и фитнесу, группы по интересам
- Сервис онлайн-записи на прием к врачу ЕМИАС
Ваше здоровье — наша цель
ВитаПортал — официальный медицинский сайт, посвященный здоровью человека. Наша основная задача – предоставить пользователю проверенную информацию, верифицированную экспертами в своих областях.
Наш сайт о здоровье создан не для практикующих врачей, а для обычных пользователей. Вся информация адаптирована и предоставлена доступным и понятным языком, медицинские термины расшифровываются. В то же время мы уделяем большое внимание проверке подлинности своих источников, которыми становятся только официальные медицинские сайты, научные медицинские журналы и практикующие врачи и эксперты.
Рекомендации и мнения, опубликованные на Сайте, включая материалы по персональной диете СлимСмайл, НЕ ЗАМЕНЯЮТ КВАЛИФИЦИРОВАННУЮ МЕДИЦИНСКУЮ ПОМОЩЬ. Обязательно проконсультируйтесь с врачом.
(томакулярная невропатия)
, MDCM, Weill Cornell Medical College
При этом заболевании нервы легко повреждаются при незначительном сдавлении, травме или повторной физической нагрузке.
В области поражения отмечаются онемение, покалывание и слабость.
При постановке диагноза важны результаты электромиографии и генетического анализа.
Пациентам следует избегать деятельности, которая вызывает симптомы, или изменять ее; одевание запястных шин и подкладывание подушечек под локти способствует уменьшению давления на пораженные нервы.
При этой наследственной невропатиипериферические нервы чувствительны к повреждениям вследствие относительно слабого давления или травмы, либо от повторяющихся движений.
Обычно болезнь начинается в юном или взрослом возрасте, но может начаться в любом возрасте. Заболеваемость одинакова для обоих полов.
Наследственную невропатию со склонностью к параличам от сдавления часто наследуется по аутосомно-доминантному типу (не связанному с полом). Это значит, что для того чтобы заболеть, ребенку достаточно получить от своих родителей только один дефектный ген.
При этой нейропатии нервы теряют миелиновую оболочку (демиелинизация) и не проводят нервные импульсы как обычно. (Миелиновая оболочка действует так же, как изоляция электрического провода, ускоряя передачу нервных импульсов).
Симптомы
Наследственная невропатия со склонностью к параличам от сдавления часто поражает нервы, которые проходят вблизи от поверхности тела, рядом с костями. Например, могут повреждаться следующие нервы:
локтевой нерв в области локтя (приводит к развитию пареза локтевого нерва);
срединный нерв в области запястья (приводит к развитию синдрома запястного канала).
Симптомы варьируются от незаметных и слабовыраженных до тяжелых и инвалидизирующих. Эпизоды могут длиться от нескольких минут до месяцев. Они могут рецидивировать, иногда затрагивая различные нервы.
После эпизода полное восстановление отмечается приблизительно у половины больных, у остальных сохраняются слабые симптомы.
Диагностика
Поскольку симптомы периодически исчезают, врачам трудно поставить диагноз наследственной невропатии со склонностью к параличам от сдавления. При постановке диагноза важны результаты электромиографии и генетического анализа.
Изредка требуется биопсия нерва.
Лечение
Больному следует избегать деятельности, которая вызывает эти симптомы.
Одевание запястных шин и подкладывание подушечек под локти
Больному следует избегать деятельности, которая вызывает эти симптомы.
Шины на запястьях и подушечки под локтями помогают уменьшить давление, предупредить повторные травмы и дать нерву время восстановиться.
Также интересно
Видео


Последнее
Компания MSD и Справочники MSD
- О нас
- Юридическая оговорка
- Разрешение
- Конфиденциальность
- Условия использования
- Лицензирование
- Обратитесь к нам
- Глобальная база медицинских знаний
- Справочник по ветеринарии (только на английском языке)
Диапазон фенотипических проявлений наследственной невропатии со склонностью к параличам - аутосомно-доминантного заболевания, характеризующегося чувствительностью периферических нервов к сдавлению. Клинические виды и механизмы развития симптомов.
| Рубрика | Медицина |
| Вид | статья |
| Язык | русский |
| Дата добавления | 07.01.2014 |
| Размер файла | 56,5 K |
- посмотреть текст работы
- скачать работу можно здесь
- полная информация о работе
- весь список подобных работ
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
HTML-версии работы пока нет.
Cкачать архив работы можно перейдя по ссылке, которая находятся ниже.
Понятие и основные причины возникновения эпилепсии как хронического психоневрологического заболевания, характеризующегося склонностью к повторяющимся припадкам, ее типы и распространенность. Эпилептический статус. Диагностика и лечение заболевания.
презентация [1,0 M], добавлен 25.11.2013
Определение и этиопатогенез невропатии. Клиническая картина заболевания. Общее понятие о параличе Белла. Невропатия тройничного, локтевого и лучевого нерва. Особенности дифференциальной диагностики заболевания. Краткая характеристика болевого синдрома.
курсовая работа [39,7 K], добавлен 28.05.2015
Описание алкогольной полинейропатии как неврологического заболевания, характеризующегося поражением функции периферических нервов. Диагностические методики полинейропатии и медикаментозное лечение. Диета, физиотерапия и массаж при лечении полинейропатии.
презентация [156,8 K], добавлен 18.02.2017
Общие сведения о заболеваниях периферической нервной системы. Причины, характеристика, лечение и особенности ухода за больными с различными видами невралгии, невропатии и полиневрита. Клинические проявления, диагностика и лечение синдрома Гийена-Барре.
контрольная работа [28,4 K], добавлен 06.11.2009
Наследственная патология органа зрения при аутосомно-рецессивном и аутосомно-доминантном типе наследования. Врождённые катаракты, нистагма, мнимый и истинный анофтальм. Глиома сетчатки глаза. Предупреждение врожденных и наследственных глазных заболеваний.
презентация [3,9 M], добавлен 27.03.2012
Травматические повреждения нервов: классификация, патоморфология. Ход процессов дегенерации и регенерации в поврежденном нерве. Синдромы и симптомы поражения периферических нервов, иннервация. Оперативное и консервативное лечение, показания, физиотерапия.
презентация [1,5 M], добавлен 21.12.2011
Дифференциация невропатий и миопатии, основные симптомы и особенности. Немиастенические синдромы при злокачественной опухоли и лучевая терапия. Беспорядочные асимметричные множественные поражения и специфические поражения отдельных периферических нервов.
реферат [20,0 K], добавлен 08.06.2009
Изучение тениоза, гельминтоза, характеризующегося поражением тонкого кишечника. Описания возбудителя заболевания. Развитие цепня свиного. Анализ симптомов, клинических проявлений и течения тениоза. Диагностика, методы лечения и профилактика гельминтозов.
презентация [1,1 M], добавлен 01.02.2015
Описание проявлений почечной недостаточности. Анамнез заболевания острым гломерулонефритом с изолированным мочевым синдромом. Общие клинические исследования проявлений заболевания у пациента. Дифференциальный диагноз, этиология и патогенез заболевания.
история болезни [53,2 K], добавлен 21.06.2015
Аллергологический, эпидемиологический, гемотрансфузионный анамнез больной. Постановка клинического диагноза невропатия левого малоберцового нерва. Составление генеалогической таблицы пациента. Обследования и лечения основного и сопутствующего заболевания.
история болезни [37,2 K], добавлен 22.04.2011

- главная
- рубрики
- по алфавиту
- вернуться в начало страницы
- вернуться к началу текста
- вернуться к подобным работам
- Рубрики
- По алфавиту
- Закачать файл
- Заказать работу
- Вебмастеру
- Продать
- весь список подобных работ
- скачать работу можно здесь
- сколько стоит заказать работу?
Работы в архивах красиво оформлены согласно требованиям ВУЗов и содержат рисунки, диаграммы, формулы и т.д.
PPT, PPTX и PDF-файлы представлены только в архивах.
Рекомендуем скачать работу.
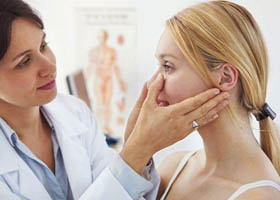
Общие сведения
Нейропатия лицевого нерва (синоним — неврит лицевого нерва, паралич Белла) представляет собой паралич/парез лицевого нерва, сопровождающийся чувствительными, двигательными и вегетативными нарушениями в зоне иннервации мимических мышц и асимметрией лица. Нейропатия лицевого нерва (НЛН) является одной из распространенных и актуальных проблем неврологии.
В первую очередь необходимо отметить, что НЛН развивается всегда лишь при поражении нервного волокна от двигательного ядра лицевого нерва до его выхода из шилососцевидного отверстия (периферический парез) и всегда на одноименной стороне в отличии от центрального пареза, который возникает преимущественно при инсульте и зачастую сочетается с парезом конечностей, развивающихся на противоположной очагу стороне (рис. ниже).
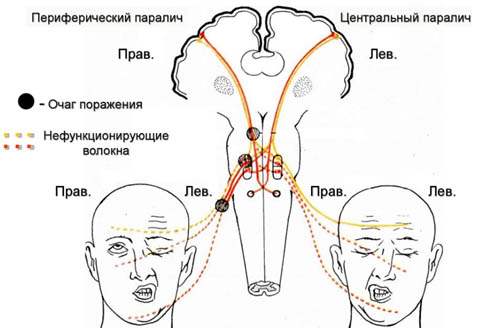
Лицевой нерв (ЛН) относится к преимущественно двигательным нервам, которые обеспечивают мимику, процессы моргания, жевания, глотания, нахмуривания. Однако в составе ствола лицевого нерва также проходят составные части промежуточного нерва — парасимпатические (секреторные) и чувствительные (вкусовые) волокна, иннервирующих слюнные железы, а также вкусовую чувствительность языка.
Относительно высокая частота поражения лицевого нерва во много обусловлена присущими ему анатомо-топографическими особенностями — нерв имеет сложный и длительный ход в узком костном канале височной кости. Наиболее уязвимым отрезком ЛН (в котором происходит его ущемление/сдавливание) является отрезок, расположенный в узком извитом канале где в случаях развития отека, обусловленного различными причинами (например, воспалением) и происходит его сдавление.
Среди разных локализаций поражения периферического отдела ЛН паралич Белла относится к наиболее часто встречаемой патологии (16-25 случаев /100 000 населения) и обусловлен развитием отека и последующей его компрессии в костном канале (туннельный синдром). Высокая ранимость ЛН в фаллопиевом канале объясняется превалированием его в поперечном сечении канала, где он занимает 40%-70% всей площади. При этом несмотря на то, что канал в отдельных местах сужается, толщина самого нервного ствола остается неизменной.
В подавляющем большинстве случаев периферический парез лицевого нерва проявляется односторонним поражением лицевого нерва. Правая/левая лицевая сторона поражаются с одинаковой частотой. На долю двусторонней невропатии ЛН приходится всего 6,2% всех его поражений. Средний возраст начала заболевания около 40 лет, но может встречаться в любом возрасте. Наименьший показатель заболеваемости отмечается у детей в возрасте до 10 лет, повышается у лиц возрастной группы 10–29 лет, стабильные показатели характерны для лиц 30–69 лет и максимальных показателей достигает в популяции больных после 70 лет.
Для заболевания характерна высокая частота осложнений (7–18% случаев), в 24,5% отмечаются рецидивирующие невропатии ЛН. Повторные невропатии по сравнению с первичными протекают более тяжело, труднее лечатся и крайне редко завершаются полным восстановлением. Нейропатия лицевого нерва, как пишут многие пациенты, посещающие специальный форум, является чрезвычайно психотравмирующей ситуацией для больных и крайне негативно отражается на психоэмоциональной сфере и физическом состоянии пациентов вплоть до развития невроза. Паралич ЛН является частой причиной длительного нарушения трудоспособности и существенно снижает качество жизни.
Патогенез
Пусковым фактором НЛН является раздражение сосудов черепно-цервикального отдела, что способствует развитию ангиоспазма позвоночной и ветвей наружной сонной артерии, что приводит к первичной ишемии корешка ЛН. Нарастающие нарушения микроциркуляции в структурах ЛН приводят к аноксическому отеку нерва. Это в сою очередь приводит к компрессии (сдавливанию) нервной ткани в лицевом (фаллопиевом) канале височной кости, нарушению нервно-мышечной проводимости, обусловленного блокадой процесса высвобождения из окончаний двигательных аксонов ацетилхолина и расстройством взаимодействия ацетилхолина с рецепторами, расположенными на постсинаптической мембране. По мере нарастания расстройств в нервной ткани развивается вторичная ишемия ЛН.
Классификация
Выделяют первичное поражение ЛН, вызванное переохлаждением и вторичное, как осложнение других заболеваний.
По этиологическому признаку выделяют:
- Паралич Белла (идиопатическая невропатия).
- Отогенные невриты (при воспалении среднего уха/сосцевидного отростка височной кости).
- Инфекционные невриты (при гриппе, герпесе, паротите, полиомиелите и др.).
- Травматические невриты (повреждение лицевого нерва).
- Ишемические (в случаях нарушении кровоснабжения нерва).
Причины неврита лицевого нерва
При периферическом характере поражения ЛН установить причины возникновения заболевания в большинстве случаев достаточно сложно. Принято считать, что причины невропатии ЛН полиэтиологичны (ишемические, отогенные, идиопатические, травматические, инфекционные и другого генеза). Как уже указывалось, паралич Белла развивается вследствие сдавления нерва в узком извитом канале височной кости, происходящего по различным причинам (воспаление, наследственная предрасположенность в виде врожденной узости канала лицевого нерва).
К провоцирующим факторам невропатии ЛН относятся переохлаждение, инфекции, сдавление нерва опухолью (невринома), травматизация костей основания черепа/лица с механическим повреждением/разрывом нервных волокон, отравления. Также невропатия может развиваться как осложнение отита, паротита, мезотимпанита, нейротропной вирусной инфекции (полиомиелита, герпеса), воспалительных процессов в головном мозге.
Симптомы
Симптомы неврита лицевого нерва определяются уровнем его поражения. Рассмотрим лишь симптоматику компрессионно-ишемического поражения ЛН (паралич Белла). Наиболее часто компрессионно-ишемическая невропатия проявляется остро развившимся парезом/параличом мимической мускулатуры в виде:
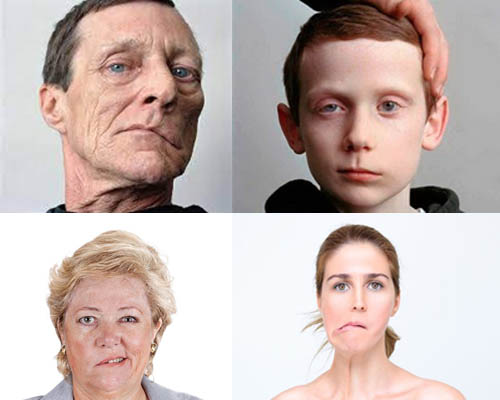
Паралич лицевого нерва начинается преимущественно внезапно. В начале заболевания у некоторых пациентов за 1-2 дня до появления двигательных расстройств или одновременно появляются умеренные/легкие боли и парестезии в области сосцевидного отростка/уха.
Анализы и диагностика
Диагноз устанавливается на основании клинической симптоматики и жалоб пациента. С целью исключения вторичной природы заболевания назначаются дополнительные инструментальные обследования (МРТ/КТ головного мозга). Для уточнения локализации поражения ЛН, степени его поражения могут назначаться электромиография и электронейрография.
Лечение неврита лицевого нерва
Вылечить парез лицевого нерва можно лишь используя комплексное лечение, включающее медикаментозную терапию, массаж, физиопроцедуры, ЛФК и специальные упражнения. Лечебные мероприятия направлены на улучшение крово/лимфообращения в области лица, нормализацию проводимости лицевого нерва, предупреждение появления мышечной контрактуры и восстановление функции мимических мышц. Лечение, в зависимости от тяжести заболевания может проводится амбулаторно или стационарно в неврологическом отделении.
Основным принципом медикаментозного лечения невропатии является снятие отека и скорейшее восстановление микроциркуляции. В остром периоде заболевания достаточно высокую эффективность имеют системные глюкокортикостероиды препараты. С этой целью проводится пульс-терапия: Метипред внутривенно капельно в течение 3 дней и далее назначается ГКС перорально в течение 5 дней, а затем доза постепенно снижает ежедневно на 5 мг. Или же назначается Преднизолон в течении 7 дней по 60-80 м/сутки с постепенной отменой за 5-6 суток. Некоторые авторы считают, что при параличе Белла более целесообразным является периневральное введение гормонов (Гидрокортизон с новокаином), что способствует более быстрой декомпрессии лицевого нерва. Параллельно назначаются диуретики — Фуросемид, Диакарб, Триамтерен.
Для снижения болевого синдрома и противовоспалительной терапии назначаются НПВС – Ксефокам, Диклофенак, Ибупрофен, Кеторолак, Зорника. Эффективны сосудорасширяющие препараты (Скополамин, Никотиновая кислота, Ксантинола никотинат). Показано назначение витаминов группы В, которые оказывают нейротропное действие, улучшают регенерацию и трофические процессы в нервной ткани, уменьшают боль (Нейромультивит, Мильгамма, Нейробион и др.).
Назначаются препараты альфа-липоевой кислоты (Тиоктацид, Берлитион, Тиогамма), способствующие восстановлению структуры нерва и купированию процессов демиелинизации.
Вне острого периода (на 7-10 сутки) для нормализации проводимости лицевого нерва проводится стимулирующая терапия — назначаются антихолинэстеразные препараты (Галантамин, Нейромидин, Ипидакрин, Аксамон). Лицевой неврит с затяжным течением требует назначения антидепрессантов — Имипрамин, Амитриптилин, Дулоксетин, Венлафаксин. В качестве дополнительного средства рекомендуется назначение оксидантов — Тиоктацид, Берлитион. Для местного обезболивания можно использовать Анестезиновую/Лидокаиновую мазь, которая наносится на болевые зоны. Если на протяжении первых 2-3- месяцев восстановление лицевого нерва в полном объеме не произошло, назначают Лидазу и препараты-биостимуляторы (ФИБС, Алоэ). При развитии контрактур показаны Мидокалм, Тегретол.
Читайте также:
