
Редкое генетическое нервное заболевание

Аквагенная крапивница

Диарея Брайнерда
В 1983 году было зафиксировано восемь вспышек диареи Брейнерда, шесть из них — в США. Но первая была все же самой крупной — за год переболело 122 человека. Имеются подозрения, что болезнь возникает после употребления парного молока — но все равно непонятно, почему она мучает человека так долго.

Тяжелые зрительные галлюцинации, или синдром Шарля Бонне
Хотя зафиксированных случаев этого заболевания немного, считается, что оно широко распространено среди людей пожилого возраста, страдающих слепотой. От 10 до 40% ослепших людей страдают от синдрома Шарля Бонне. К счастью, в отличие от других перечисленных здесь заболеваний, симптомы тяжелых зрительных галлюцинаций исчезают сами по себе после года или двух, так как мозг начинает приспосабливаться к утрате зрения.

Электромагнитная сверхчувствительность

Синдром скованного человека

Кивательный синдром

Аллотриофагия

Английский пот

Болезнь Перуанского метеорита
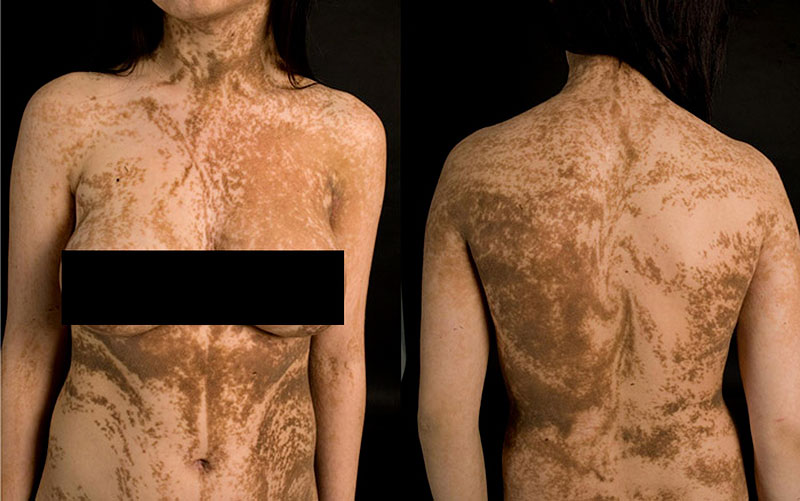
Линии Блашко

Болезнь куру, или смеющаяся смерть
Болезнь распространялась через ритуальный каннибализм, а именно поедание мозга болевшего этим заболеванием. С искоренением каннибализма куру практически исчезла.
Синдром циклической рвоты

Синдром синей кожи, или акантокератодермия

Болезнь Моргеллонов

Болезнь двадцатого века

Хорея

Прогерия (Progeria), синдром Гетчинсона — Гилфорда

Порфирия
Причины возникновения этого заболевания до сих пор изучены недостаточно. Известно, что оно наследственное и связано с неправильным синтезированием красных кровяных телец. Многие ученые склоняются к тому, что в большинстве случаев оно возникает в результате инцеста.

Синдром войны в заливе

Синдром прыгающего француза из штата Мэн
10 опасных неврологических заболеваний, передающихся по наследству

Зачастую это заболевание дает о себе знать в пожилом возрасте. Оно проявляется стремительным ухудшением двигательной активности, тремором рук и ног, замедленными реакциями и движениями. Кроме того, при данном заболевании наблюдается развитие депрессии, иногда тяжелой, и различные нарушения интеллекта.
Слабоумие, как ее еще называют, развивается у людей преклонного возраста. Первые признаки болезни Альцгеймера — ухудшение кратковременной памяти, спутанность сознания. Позже появляется раздражительность и агрессия, а со временем нарушается способность говорить и воспринимать речь, происходит затухание сознания, что ведет к потере функций организма и смерти.
Боковой амиотрофический склероз
Это неизлечимая прогрессирующая болезнь центральной нервной системы, этиология которой пока до конца не разгадана. При данном заболевании верхние и нижние двигательные нейроны в головном мозге подвергаются дегенеративному поражению, из-за чего происходит паралич и атрофия мышц. В результате всех этих процессов пациент стремительно умирает от инфекций дыхательных путей или отказа дыхательной мускулатуры.
Это одно из самых сложных заболеваний головного мозга, которое также запечатлевается геноммом и передается новым поколениям. Заболевание характеризуется психическими расстройствами и неконтролируемыми импульсивными движениями. При тяжелых стадиях заболевания появляются такие симптомы, как галлюцинации, беспричинные истерики и приступы агрессии, полный распад личности, а речь при этом становится бессмысленной и неразборчивой.
При данном заболевании происходит наращивание липопигментов в клетках головного мозга, глаз, кожи и в других тканях. Главными симптомами заболевания являются несогласованность мышц, мышечные спазмы, ухудшение зрения или слепота, снижение тонуса мышц, нарушения психики и умственная отсталость.
Это хроническое расстройство нервной системы и психики, которое может показать себя как в детстве, так и во взрослом возрасте. В большинстве случаев пациенты с эпилепсией не имеют интеллектуальных нарушений и деструктивных расстройств ЦНС. Основная опасность эпилепсии состоит в приступах, которые могут произойти в любое время и стать причиной смерти.
Мышечная дистрофия Беккера
Данные заболевания характеризуются нарушением работы произвольной мускулатуры. Так, пациент не имеет двигательного контроля по причине поражения нервов, нейронов спинного мозга или непосредственно мышц. Признаками таких заболеваний являются слабость и атрофия мышц, онемения, судороги и спазмы, боли и покалывания в мышцах. Главная опасность болезни заключается в поражении дыхательных и глотательных функций.
Это заболевание еще называют подострой некротизирующей энцефаломиелопатией. Зачастую заболевание проявляется в раннем детстве – 1-2 года. Симптомами болезни являются атрофия зрительных нервов, задержка психомоторного развития, слабость в мышцах, паралич и тугоухость.
Данное заболевание поражает нейроны головного мозга. Оно проявляется еще в младенческом возрасте – до полугода. При данном заболевании происходит задержка развития, потеря мышечного тонуса, проблемы с питанием и патологическое увеличение головы. В большинстве случаев (порядка 70% детей) с болезнью Кэнэвэна не доживают до 10-летнего возраста.
Это заболевание связано с распадом мыслительных и эмоциональных процессов. Его симптомами являются галлюцинации, бред, нарушение речи и работоспособности. У пациентов с шизофренией повышается риск развития тяжелых депрессий, которые увеличивают риск увлечения наркотиками или алкоголем, а также суицидальных наклонностей.
Список редких генетических заболеваний и расстройств от А до Я
Генетические заболевания присутствуют на протяжении всей жизни человека, некоторые из которых появляются очень рано. Они приводят ко многим хроническим заболеваниям, которые не поддаются лечению. Вот редкие генетические заболевания и расстройства, которые наблюдаются у людей.
Генетические заболевания или расстройства возникают из-за аномалий в генетическом составе человека. Такие аномалии могут быть вызваны незначительными, значительными изменениями или мутациями в одном или нескольких генах, хромосомными аберрациями и редко из-за мутаций в нехромосомной ДНК митохондрий. Генетические заболевания или расстройства могут быть или не быть наследственными. Они могут быть рецессивными или доминирующими по природе.
Редкая болезнь в одной части мира не может быть редкой в другой. В Соединенных Штатах критерии редких заболеваний определены в Законе о редких заболеваниях 2002 года. Он определяет такие заболевания строго в соответствии с его распространенностью, а именно «любое заболевание или состояние, которым страдают менее 200 000 человек в Соединенных Штатах, или около 1 в 1500 году. "Существует около 6000 известных генетических заболеваний, многие из которых являются дегенеративными, изнурительными и часто смертельными. В этой статье давайте узнаем о некоторых более редких формах генетических заболеваний и расстройств.
Синдром Айкарди
Синдром Айкарди - это очень редкое генетическое заболевание, характеризующееся недоразвитостью или отсутствием мозолистого тела, структуры, разделяющей левую и правую половину мозга. Оказалось, что он влияет только на девочек, так как считается, что он вызван дефектом Х-хромосомы. Это затрагивает 1 из 100 000 - 150 000 человек в Соединенных Штатах. Младенцы, рожденные с этим расстройством, кажутся нормальными до возраста от 3 до 5 месяцев, и затем начинают проявляться некоторые ключевые симптомы этого расстройства.
Симптомы включают аномалии сетчатки и инфантильные спазмы, приводящие к судорогам. Люди, затронутые этой болезнью, имеют серьезные проблемы с развитием.
Лечение не существует, нет лекарства от этого расстройства. Лечение обычно включает управление судорогами и оказание поддержки пострадавшим людям за счет задержки в развитии.
Синдром Алагилла
Синдром Алагилла - это редкое генетическое заболевание, которое поражает печень, почки, сердце и другие органы тела. Симптомы, связанные с этим синдромом, обычно отмечаются в первые годы жизни. Это затрагивает приблизительно 1 из 70 000 новорожденных. Он наследуется как аутосомно-доминантный признак, и тяжесть симптомов может варьироваться от человека к человеку. Нарушение работы печени вызвано аномалиями в желчном протоке (например, меньше или отсутствует, узкий или неправильно сформированный), что приводит к накоплению желчи в печени и, таким образом, к ее повреждению.
Симптомы включают желтоватый оттенок на коже и белках глаз, накопление холестерина под кожей, зуд и т. Д. Люди, страдающие этим расстройством, также сталкиваются со многими проблемами с сердцем. У них различимые черты лица - выступающий лоб, острый подбородок и глубоко посаженные глаза.
Лечение от этого синдрома нет, как и лекарства. Однако коррекционная хирургия, направленная на функционирование сердца, печени и почек, в некоторой степени помогает.
Алкаптонурия
Алкаптонурия, также известная как болезнь черной мочи, вызвана нарушением метаболизма тирозина в организме. Это затрагивает приблизительно 1 из 250 000 - 1 миллион человек во всем мире. Это аутосомно-рецессивное состояние, характеризующееся накоплением в крови алкаптона или гомогентизиновой кислоты (токсичного побочного продукта тирозина), которая выделяется с мочой. Моча таких пациентов становится черной при воздействии воздуха. Присутствие избытка алкаптона может привести к остеоартриту, болезням сердца, камням в почках и камням простаты у мужчин.
Симптомы: Некоторые из симптомов алкаптонурии включают потемневшую кожу и пигментированную склеру (белая часть глаза).
Лечение: Особого лечения не было установлено. Однако было отмечено, что помогает употребление продуктов, богатых витамином С. Также желательно избегать диет, богатых тирозином и фенилаланином.
Синдром альстрёма
Синдром Альстрёма - это редкое аутосомно-рецессивное заболевание, вызывающее полиорганную дисфункцию. Это одно из самых редких известных генетических заболеваний, в истории которого известно всего около 500 случаев.
Симптомы: Показания включают детское ожирение, нейросенсорную тугоухость и нарушение зрения. Это также может привести к гиперинсулинемии, гипертриглицеридемии, раннему развитию диабета 2 и глухоте. Этот синдром также может вызывать некоторые опасные для жизни медицинские осложнения, связанные с поражением печени, сердца, легких и т. Д.
Лечение: от этого синдрома нет лекарства; Тем не менее, лекарства могут быть предоставлены для лечения конкретных симптомов синдрома.
Аперт синдром
Аперт синдром является редким генетическим заболеванием, которое проявляется при рождении. Это приводит к искажению формы черепа и лица, а иногда руки и ноги перепончатые. Это влияет на 1 из 65 000 до 88 000 новорожденных. У людей, пораженных этим синдромом, кости черепа преждевременно срастаются, что называется краниосиностозом, в то время как мозг продолжает развиваться внутри аномального черепа, вызывая давление на череп и лицо, что приводит к его искажению.
Симптомы: это также приводит к плохому интеллектуальному развитию человека, потере слуха, частым инфекциям уха и пазухи и низкому росту. В большинстве случаев синдром возникает в результате случайной генной мутации, а в некоторых редких случаях он наследуется как аутосомно-доминантный признак.
Лечение - это не излечивается, но хирургическое вмешательство может до некоторой степени вылечить некоторые из его симптомов.
Болезнь Баттена
Болезнь Баттена - это редкое аутосомно-рецессивное заболевание, которое в основном поражает нервную систему и начинается в детском возрасте. По оценкам, это происходит в каждых 2-4 из каждых 100 000 человек. Болезнь Баттена характеризуется накоплением пигментов, называемых липофусцинами, в клетках организма.
Симптомы. Симптомы обычно появляются в возрасте от 5 до 10 лет, когда у нормального ребенка внезапно появляются проблемы со зрением и судороги. С течением времени симптомы ухудшаются, что приводит к потере двигательных способностей, умственной отсталости, потере зрения и т. Д.
Лечение. От этого состояния не существует лекарства, и к 20 годам оно обычно приводит к летальному исходу.
Синдром Бардета-Бидля
Синдром Бардета-Бидля - это редкое генетическое заболевание, поражающее несколько органов. Поражает от 1 из 140 000 до 1 из 160 000 новорожденных. Признаки и симптомы могут отличаться среди людей, которые страдают от синдрома, даже среди членов семьи.
Симптомы: для него характерны нарушения зрения, ожирение, аномалии почек, проблемы развития, лишние пальцы рук и ног, нарушение двигательных навыков и т. Д. Это расстройство носит рецессивный характер.
Лечение: лечения от этой болезни нет, и обычно лечение концентрируется на конкретных симптомах.
Болезнь Камурати-Энгельмана
Болезнь Камурати-Энгельмана является разновидностью дисплазии кости. Это редкое аутосомно-доминантное генетическое заболевание. На сегодняшний день во всем мире зарегистрировано только 200 случаев заболевания.
Симптомы: для него характерны утолщенные кости, что приводит к хронической боли. Кости черепа также утолщаются, что приводит к давлению на мозг, что приводит к различным неврологическим проблемам. Люди, страдающие от этого, также жалуются на повышенную утомляемость, слабость, головную боль и мышечные спазмы.
Лечение: от этого состояния нет лекарства, но его можно частично лечить. Противовоспалительные и иммунодепрессивные агенты, такие как глюкокортикостероиды, оказались полезными в некоторых случаях. Также рекомендуются альтернативные методы лечения, такие как массаж и тепловая терапия в сочетании с медикаментозным лечением.
Синдром Карпентера
Синдром Карпентера - это редкое врожденное заболевание, характеризующееся неправильной формой головы, лица, пальцев рук и ног из-за преждевременного сращения костей. Это аутосомно-рецессивное заболевание с примерно 100 зарегистрированными случаями, известными до настоящего времени. По оценкам, это влияет на 1 из 1000 000 человек.
Симптомы: Основные симптомы включают головку странной формы, слитые цифры, ожирение, низкий рост и снижение умственных способностей. 50% детей с этим расстройством имеют порок сердца, который осложняет ситуацию.
Лечение: Лечение обычно состоит из ряда поэтапных операций, направленных на исправление пороков развития костей на ранних этапах жизни. Это состояние похоже на синдром Аперта и синдром Пфейффера.
Семейные идиопатические базальные ганглии кальцификации
Ранее известный как синдром Фара, это редкое генетически доминирующее заболевание. Характеризуется аномальными отложениями кальция в базальных ганглиях и коре головного мозга. Это области управления движением мозга.
Симптомы: Ключевыми симптомами являются неуклюжесть, усталость, неустойчивая осанка, мышечные спазмы, неконтролируемые движения и деменция. Люди могут быть затронуты этим синдромом в любое время их жизни; однако, это более распространено в возрастной группе 40-50 лет. Известно, что только 60 семей страдают от этого синдрома в медицинской литературе.
Лечение: от этого состояния нет лекарства. Лечение обычно симптоматическое.
Наследственный ангионевротический отек
Наследственный ангионевротический отек, также известный как болезнь Квинке, вызван нарушениями функции белка, называемого ингибитором С1. Мутации в гене ингибитора C1 (C1NH) приводят к HAE. Это влияет на кровеносные сосуды, что приводит к проблемам иммунной системы. По оценкам, это происходит в 1 из 50000 человек.
Симптомы: Общие симптомы наследственного ангионевротического отека включают отек рук, ног, глаз и горла, боли в животе и закупорку дыхательных путей.
Лечение: Лечение обычно проводится после приема лекарств, гормонального лечения и введения обезболивающих препаратов.
Болезнь Хантингтона
Болезнь Хантингтона является аутосомно-доминантным генетическим заболеванием и более распространена в зрелом возрасте. Со временем симптомы усугубляются, и пострадавшему человеку требуется постоянный уход. Это условие влияет на 1 из 10000 в Соединенных Штатах. Ожидаемая продолжительность жизни после начала заболевания составляет 15-25 лет.
Симптомы: Показатели включают непроизвольные движения, потерю двигательных способностей, когнитивные трудности и эмоциональные проблемы.
Лечение: лечения не существует, но лекарства могут облегчить определенные симптомы, связанные с расстройством.
Синдром Меккеля
Синдром Меккеля - чрезвычайно редкое фатальное генетическое заболевание. Он наследуется по аутосомно-рецессивному типу и поражает примерно 1 из 13 250-140 000 человек во всем мире. Из-за его серьезных симптомов люди обычно умирают во время родов или вскоре после рождения. Это чаще встречается в Бельгии и Финляндии, где примерно 1 из 3000-9000 человек страдает от этого.
Симптомы : у него тяжелые признаки и симптомы, включая уродливую центральную нервную систему, многочисленные заполненные жидкостью кисты в почках, дефекты легких, печени, полидактилию.
Лечение : может быть рекомендовано восстановление сердца или нейрохирургическое вмешательство при энцефалоцеле.
Мукополисахаридоз VI
Мукополисахаридоз VI - это генетическое заболевание, которое встречается у 1 из 300 000 новорожденных. Это вызвано дефицитом активности N-ацетилгалактозамин-4-сульфатазы (арилсульфатазы В), которая ответственна за катаболизм сложных углеводов (полисахаридов). Это приводит к накоплению дерматансульфата в некоторых органах, таких как скелет, легкие, клапаны сердца, селезенка и печень.
Симптомы: Симптомы включают низкий рост, умственную отсталость, респираторные заболевания, глухоту и слепоту.
Лечение: Лечение проводится после заместительной энзимотерапии и регулярной медицинской помощи.
Синдром Пфейффера
Синдром Пфайффера связан с мутацией рецептора фактора роста фибробластов, который важен для нормального развития костей. Это редкое аутосомно-доминантное генетическое заболевание, поражающее 1 человека на 100 000 человек. Характеризуется аномальным слиянием костей черепа, что приводит к деформации головы и лица. Это также может повлиять на кости ног и рук.
Симптомы: Видимые симптомы этого синдрома: необычно широкий лоб, выпученные глаза, клюв носа, короткие широкие пальцы рук и ног. В некоторых случаях также видна перепонка цифр. Индивидуум, страдающий от этого синдрома, также сталкивается с потерей слуха и зубными проблемами.
Лечение: поэтапные черепно-лицевые операции обычно проводятся в первые месяцы жизни, чтобы исправить деформацию костей.
Прогерия
Прогерия - это редкое генетическое заболевание, которое встречается у 1 из 8 000 000 живорожденных. Это генетическое заболевание, вызванное новой мутацией в гене и, как правило, не наследуемое. Это вызывает быстрое старение у детей, и в результате люди, затронутые этой болезнью, умирают в возрасте от 13 до 20 лет.
Симптомы: Ключевыми симптомами прогерии являются проблемы роста на первом году жизни, морщинистое маленькое лицо, большая голова, потеря волос, бровей и ресниц, плохое зрение и снижение моторных навыков. Генетический тест на мутацию LMNA может подтвердить, есть ли у человека прогерия.
Лечение: от этой болезни нет лекарства. Все предлагаемые варианты лечения, как правило, носят поддерживающий характер и зависят от симптомов.
Синдром Ретта
Синдром Ретта - это редкое генетическое нарушение развития нервной системы. Это аутосомно-доминантное Х-сцепленное расстройство, которое чаще встречается у женщин, чем у мужчин. Это затрагивает 1 из 10000 человек. Симптомы обычно появляются после 6-18 месяцев жизни.
Симптомы: они включают проблемы с дыханием, сенсорные проблемы, неустойчивую походку, неспособность к обучению, судороги, социальную неловкость или безответственность. Симптомы часто неверно интерпретируются как аутизм и церебральный паралич.
Лечение: от этого синдрома нет лекарства; однако исследования в этой области продолжаются.
Дефицит рибозо-5-фосфат-изомеразы
Дефицит возникает из-за мутации в энзиме рибозо-5-фосфат-изомеразы, которая играет жизненно важную роль в пентозофосфатном пути. Только с одним диагностированным случаем это расстройство считается самым редким. МРТ показала высокий уровень арабита и рибита, что указывает на врожденную ошибку метаболизма полиолов. У пациента были такие симптомы, как болезнь белого вещества и невропатия неизвестного происхождения. Причину этого состояния еще предстоит полностью понять. Есть несколько теорий, делающих раунды, но этот недостаток все еще изучается.
Болезнь Сандхоффа
Болезнь Сандхоффа - это редкое генетическое нарушение накопления липидов, которое разрушает нервные клетки головного и спинного мозга. Это вызвано из-за дефицита функциональных бета-гексозаминидаз A & B, которые приводят к отложению определенных липидов в мозге и других органах тела. Это аутосомное расстройство затрагивает 1 из 300 000 человек. Из 3-х типов этого синдрома классический инфантильный является самым летальным.
Симптомы: У пораженных детей проявляются мышечная слабость, потеря двигательных навыков, страдают потерей слуха, параличом, потерей зрения, пятнами красной вишни, умственными нарушениями и обычно выживают до возраста 3-4 лет.
Лечение: Лечение этого состояния, как правило, является поддерживающим.
Туберозный склероз
Туберозный склероз ранее был известен как болезнь Борнвилля. Это редкое генетическое заболевание, которое поражает примерно 1 из 10 000 человек во всем мире. Около 80% случаев связаны с мутациями в двух специфических генетических локусах - TSC1 и TSC2. Туберозный склероз вызван ростом опухолевидных опухолей головного мозга, легких, сердца и почек.
Симптомы: Общие симптомы включают поражения кожи в области носа и щек, периунгулярную фиброму, эпилептические припадки, поведенческие проблемы, заболевания легких и почек и умственную отсталость.
Лечение: Симптомы эпилептических припадков можно лечить хирургическим путем, удаляя клубни, которые отвечают за эпилепсию. Проводятся исследования, чтобы найти лекарственную терапию для этого заболевания.
Болезнь Вольмана
Болезнь Вольмана - это редкое генетическое заболевание, поражающее примерно 1 из 35 000 человек. Это аутосомно-рецессивное заболевание, вызванное дефицитом лизосомально-кислой липазы (LAL). LAL - это фермент, необходимый для расщепления определенных липидов в клетках. У людей, страдающих от этого состояния, вредное количество липидов накапливается в печени, селезенке, лимфатических узлах, костном мозге и других частях тела. Симптомы обычно проявляются в первые несколько недель жизни.
Симптомы: к ним относятся проблемы с кормлением, постоянная рвота, диарея, увеличение печени и селезенки, плохое увеличение веса и т. д.
Лечение : от этого состояния не существует лекарства; Однако существуют методы лечения, которые являются симптоматическими и поддерживающими.
Синдром Циммерманна-Лабанда (ZLS)
Только с 44-мя известными случаями синдром Циммермана-Лабанда нам известен. Он является очень редким врожденным аутосомно-доминантным заболеванием.
Симптомы: для него характерен фиброматоз десен (крупные десны), отсутствие или недоразвитие ногтей конечных фаланг пальцев рук и ног, аномальный нос и ухо и грубый внешний вид лица.
Лечение: Лечение этого синдрома обычно симптоматическое и поддерживающее.
Генетические заболевания и расстройства являются серьезной проблемой для медицинских исследователей во всем мире. Исследования по клонированию генов, генной терапии и подавлению генов, ответственных за возникновение генетических заболеваний и замену ферментов, все еще находятся в поисках способов лечения редких генетических заболеваний и расстройств.
Многих болезней можно избежать, поддерживая здоровый образ жизни или сделав вакцинацию. Этого вполне хватает, чтобы неприятные симптомы обошли стороной. Но не от всего на свете существует профилактика. Редкие наследственные заболевания преследуют человека как ни защищайся. Некоторые из таких болезней можно отсрочить, другие — нет.
Генетический код, который мы получаем от отца с матерью, несет в себе не только полезную информацию. Иногда случается так, что гены с мутацией получает ребенок вполне здоровых родителей. А впоследствии оказывается, что такие же отклонения были у бабушек или прадедушек. Сегодня поговорим о самых редких заболеваниях в мире.

Прогерия
Ускоренное во много раз старение тела, тремор и атеросклероз в возрасте 5-6 лет. Это последствия прогерии или синдрома Хатчинсона-Гилфирда. Мутация гена, отвечающего за нормальное деление клеток приводит к тому, что ребенок в совсем юном возрасте получает организм старика со всеми неприятными болезнями пожилого возраста.
Диагностируют это заболевания в возрасте примерно 2 лет. Для него характерны:
- Заметная задержка роста. Также может быть очень позднее прорезывание зубов.
- Физические признаки старения. Такие, как дряблая кожа, облысение, плохая работа суставов.
- Аномалия черепно-лицевых костей. Диспропорция в сторону увеличения верхней части черепных костей.
Прогерия на сегодняшний день — это неизлечимый дефект, и люди, которые им страдают, очень редко доживают до совершеннолетия. Медики занимаются углубленным исследованием данного отклонения, надеясь найти ответ, как помешать измененному гену забирать жизни детей.
Нарколепсия-катаплексия
Патологическая дневная сонливость может встречаться у людей разного возраста. Нарколепсия характерна внезапным наплывам усталости и слабости с которыми невозможно бороться. Больной способен мгновенно уснуть в любом месте и положении. Вместе с этим, появляются и следующие симптомы:
- сонный паралич.
- галлюцинации.
- катаплексия.
О последнем проявлении стоит поговорить отдельно. Внезапная потеря тонуса мышц, или катаплексия, является неизменным спутником ПДС. Провоцирует такое оцепенение проявление достаточно сильной эмоции, такой как гнев или радость. Слабость наблюдается как локально, так и во всем теле.
Если говорить о генетической составляющей этой болезни, то чаще всего она вызвана аутоиммунным разрушением нейронов в гипоталамусе. Наследственная передача нарколепсии вызывает много споров у докторов медицинских наук. В наше время нарколепсию-катаплексию лечат при помощи активных препаратов для усиления бодрствования и некоторыми антидепрессантами.

Синдром Юнера Тана
К категории “Самые редкие генетические заболевания” относят необычное отклонение, при котором люди не способны передвигаться на двух конечностях. Турецкий биолог Юнер Тан, который открыл это заболевание, считает, что таким образом эволюция повернула вспять. Однако, многие ученые с ним не согласны.
В семье, где обнаружили проявление синдрома всего 19 человек. И только 5 из них не могут стоять и ходить ровно. Изучая такую аномалию, ученые стали находить в разных уголках планеты схожие случаи. Сопровождать болезнь могут еще и различные умственные отклонения. Но это нельзя считать основным признаком, поскольку некоторые из носителей синдрома все же вполне полноценно контактируют с другими людьми.
На сегодняшний день ведутся различные исследования, которые направлены на изучение этого явления. Семья, в которой обнаружены люди с синдромом Юнера Тана известна на весь мир. Но точного определения, почему они получили эту особенность, нет. Равно как и не выявлен ген, несущий подобную мутацию.
Бородавочная дисплазия
Аутосомно-рецессивное генетическое заболевание, которое влияет на кожу человека и способствует появления огромных наростов, называется бородавочной эпидермодисплазией. Больных этим отклонением называют “человек-дерево” по причине схожести наростов с корнями и корой. На сегодняшний день в мире насчитывают всего несколько десятков людей с данной мутацией.
Аномальная восприимчивость к вирусу папилломы приводит к тому, что чешуйчатые папулы разрастаются до невероятных размеров. Чаще всего эти образования появляются на руках и ногах больного. Однако наросты могут быть также на лице, особенно на ушных раковинах, или теле.
Современная медицина может предложить только один выход — хирургическое вмешательство. Однако, спустя некоторое время наросты возвращаются, и операцию нужно проводить снова. Ученые занимаются изучением генетической мутации, но положительных результатов это пока не дает.
Прогрессивная липодистрофия
Редкие генетические заболевания человека часто ставят ученых в тупик. Так случилось и с прогрессивной липодистрофией. Вследствие мутации отдельных генов, носитель страдает от быстрого разрушения подкожной жировой ткани. В результате этого кожа начинает провисать и на ней появляются морщины.
Человек страдающий липодистрофией выглядит намного старше своего возраста. Чаще всего этому изменению подвержены женщины, но зафиксировано и несколько случаев у мужчин. Интересно, что медицина до сих пор не выявила причину этого отклонения.
Если говорить о лечении, то в случае с прогрессивной липодистрофией оно может быть только симптоматическое. Врачи помогут облегчить болезненность кожи и при помощи косметических средств сделают подтяжку лица. Но такой подход помогает только временно. 
Гипертрихоз
Тело человека почти полностью покрыто волосами, и это считается абсолютной нормой. В некоторых местах это едва заметный пушок, в других — достаточно жесткие и толстые стержневые волосы. Генетическое отклонение под названием гипертрихоз вызывает интенсивный рост волос на тех участках, где это не предусмотрено природой.
Выделяют несколько видов этого отклонения:
- Врожденный пушковый гипертрихоз, когда волосы после рождения не меняются, а продолжают расти.
- Приобретенный пушковый гипертрихоз. В этом случае из волосяных фоликул растут не пушковые или терминальные волоски, а зародышевые волосы.
- Лекарственный гипертрихоз, когда рост волосяного покрова активизируется под действием отдельных препаратов.
- Симптоматический гипертрихоз. Волосы растут как следствие отдельных болезней.
- Травматический гипертрихоз. На месте шрамов и рубцов наблюдается усиленный рост волос.
Это генетическое отклонение поддается лечению в трихологических центов. Стоит помнить, что самостоятельное удаление нежелательных волос приводит только к усилению роста. Лечение проходит комплексно, используя несколько действенных методов.
Конгенитальная миотония
Микроцефалия
Пигментная ксеродерма
Достаточно тяжелое наследственное заболевание, которое характеризуется аномальной чувствительностью кожи к Уф-лучам. Пигментная ксеродерма влечет за собой различные воспалительные процессы кожи и образование онкологических клеток. Нередко наличие этой генной мутации приводит к смерти, потому крайне важно постоянное врачебное наблюдение.
Для страдающих этим заболеванием солнечные лучи являются действительно убийственными. Симптомами являются покраснения и образование пузырьков на участках кожи, куда попал солнечный свет. Нередко отклонение влечет за собой фото и светобоязнь. Излечиться от пигментной ксеродермы до сих пор нельзя. Но при соблюдении всех мер предосторожности, вполне реально прожить достаточно долго.
Географический язык
Одна из самых безобидных болезней, которая может быть вызвана наследственностью — это десквамативный глоссит или географический язык. Она вызывается воспалительными процессами, в результате которых начинают страдать вкусовые рецепторы и появляются трещины на поверхности языка. Симптомы болезни могут быть разные:
- Образование красных рельефных пятен на языке.
- Повышенная чувствительность к сильным вкусовым раздражителям, горячей и холодной воде.
- Дискомфорт и жжение в ротовой полости.
Лечения как такового болезнь не требует, поскольку не является опасной для здоровья. Симптомы легко убрать при помощи антисептиков и регенерирующих препаратов. Нередко для устранения проявлений глоссита используют различные народные средства, которые также помогают успешно бороться с проявлениями симптомов.
Ихтиоз арлекина
В большинстве своем очень редкие заболевания человека в результате наследственных мутаций не несут особых угроз для жизни. Однако среди действительно опасных отклонений ихтиоз арлекина занимает едва ли не первое место. В целом ихтиоз — это нарушение обмена веществ в организме, в результате которого кожа грубеет и покрывается чешуйками.
Редчайшее генетическое отклонение характеризуется очень толстой кожей, покрытой трещинами. При этом аномальным изменениям поддаются также веки и рот. Уход за младенцем с этой болезнью — это постоянные купания и увлажнения кожи. Но даже с самым тщательным уходом дети с ихтиозом арлекина живут всего несколько месяцев. Только в 2% случаев больным удается протянуть до 10-12 лет. Врачи до сих пор не определили, может ли существовать действенное лечение этого ихтиоза.
Читайте также:
