
Сенсорно вегетативная невропатия iv типа
Автор - д.м.н. Елена Леонидовна Дадали Письмо Автору
НАСЛЕДСТВЕННАЯ СЕНСОРНО-ВЕГЕТАТИВНАЯ НЕЙРОПАТИЯ ТИП 4, ВРОЖДЕННОЕ ОТСУТСТВИЕ ЧУВСТВИТЕЛЬНОСТИ К БОЛИ С АНГИДРОЗОМ (OMIM: 256800)
Заболевание впервые описано Swanson с соавт., в 1963 году у двух братьев.
Клинические проявления возникают с рождения и характеризуются отсутствием болевой и температурной чувствительности, эпизодами необъяснимой гипертермии и тахипноэ, отсутствием потоотделения, даже при проведении провокационных проб. Длительные периоды гипертермии, не купирующиеся известными жаропонижающими препаратами могут приводить к смерти больного. Описаны больные с наличием фебрильных судорог и нарушения функции сфинктеров. Отсутствие болевой чувствительности часто приводит к возникновению травматических повреждений, (а у больных в раннем возрасте - самоповреждений) языка, губ, пальцев кистей, а также ожогов. При осмотре больных в старшем возрасте обращает на себя внимание наличие множественных рубцов, особенно в области лица. Отмечается значительное снижение чувствительности роговицы, ее помутнение и образование плохо заживающих язв. Слезотечение у больных, как правило, уменьшено и не увеличивается при введении неостигмина.
У ряда больных выявлено снижение интеллекта.
Характерным электромиографическим показателем является отсутствие или снижение амплитуды сенсорного потенциала действия.
При морфологическом исследовании отмечается почти полное отсутствие афферентной части периферических нервов, обеспечивающей проведения болевых и температурных импульсов. При прижизненном исследовании кожных ветвей лучевого нерва Rafel с соавт., в 1980 году обнаружили практически полное отсутствие как миелинизированных, так и немиелинизированных волокон, что расценивалось авторами как порок развития периферического нерва.
Тип наследования аутосомно-рецессивный.
Ген заболевания, обозначаемый как NTRK1 (ген нейротрофинового рецептора тирозинкиназы 1 типа) локализован на хромосоме 1q 21-22 , содержит 17 экзонов и занимает 25 тысяч п.н.. Наиболее частым типом мутационного повреждения гена являются точковые мутации, которых к настоящему времени идентифицировано 11. В редких случаях причиной заболевания может быть однородительская дисомия 1 хромосомы.
Заболевание является аллельным вариантом медулярной тиреоидной карциномы.
Показано, что нейроторофины и их рецепторы играют важную роль в регуляции процессов роста и диференцировки центральной и перифирической нервной системы. Биологическая роль нейротрофинов может быть различной. Так предшественники зрелых форм усиливают апоптоз нейронов, а зрелые формы обеспечивают процессы их выживаемости.
Мутации в гене приводят к нарушению функционирования как экстрацелюлярного домена, связанного с фактором роста нервов - полипептидом вовлеченным в регуляцию роста и дифференцировки вегетативных и сенсорных нейронов, так и интрацелюлярного домена, обеспечивающего сигнальную трансдукцию.
Возможна дородовая диагностика.
- Indo, Y.; Tsuruta, M.; Hayashida, Y.; Karim, M. A.; Ohta, K.; Kawano, T.; Mitsubuchi, H.; Tonoki, H.; Awaya, Y.; Matsuda, I.: Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Na- ture Genet. 13: 485-488, 1996.
- Swanson, A. G.: Congenital insensitivity to pain with anhidrosis: a unique syn- drome in two male siblings. Arch. Neurol. 8: 299-306, 1963.
- Swanson, A. G.; Buchan, G. C.; Alvord, E. C., Jr.: Absence of Lissauer's tract and small dorsal root axons in familial, congenital, universal insensitivity to pain. Trans. Am. Neurol. Assoc. 88: 99-103, 1963.
Наследственные периферические сенсорные невропатии редки, и окончательный диагноз достаточно сложен. Из-за противоречивой терминологии классификация не упорядочена (Axelrod и Pearson, 1984), а некоторые диагностические тесты могут быть недоступны за пределами специализированных центров. Общее лечение сенсорных невропатий обсуждается Klein и Dyck (2005а).
а) НСВН I (сенсорная корешковая невропатия, acropathie ulceromutilante). НСВН I отличается от всех остальных НСВН тем, что при ней симптомы появляются поздно, обычно после первого десятилетия жизни, а не в грудном возрасте. Наследование аутосомно-доминантное. Ген классической формы, ген длинноцепочечной субъединицы 1 серин-пальмитоилтрансферазы (SPTLC1), участвует в биосинтезе фосфолипидов и картирован на 9q22.1-q22.3 (Dawkins et al., 2001). Патоморфологическое исследование показывает заметное сокращение числа немиелинизированных волокон. Уменьшение толстых и тонких миелинизированных волокон меньше, но достаточно выраженное.
Ганглии задних корешков и задние корешки спинного мозга, связанные с нижними конечностями, дегенерированы. Симптомы появляются в старшем детском возрасте или пубертатном периоде в виде прогрессирующей потери чувствительности в нижних конечностях, быстро осложняющейся эпизодами целлюлита и трофическими язвами на ногах. Может встречаться спонтанная пронизывающая боль. При этом происходит потеря болевой и температурной чувствительности с сохранением тактильной чувствительности. Позднее возможно полное исчезновение чувствительности и поражение дистальных отделов верхних конечностей. Часто присутствует нейросенсорная тугоухость. В конечном итоге нередка перонеальная слабость. Моторные скорости проведения по нерву умеренно снижены, а сенсорные потенциалы действия отсутствуют. Течение заболевания медленно прогрессирующее. НСВН I гетерогенна. Мутации RAB7 могут вызвать подобную картину или более типичный фенотип синдрома Шарко-Мари-Тута (ШМТ 2В).
б) НСВН II (врожденная сенсорная невропатия). Заболевание представляет собой аутосомно-рецессивное состояние с врожденным или ранним началом. У большинства пациентов клинически проявляется отсутствие всех видов болевой чувствительности, приводящее к ожогам и калечащим повреждениям губ или кончиков пальцев и к безболезненным переломам, особенно в метатарзальной области. Тактильная чувствительность также заметно нарушена. Зоны нормальной чувствительности сохраняются у некоторых пациентов с преобладающим поражением конечностей и лица. При растяжении мочевого пузыря может нарушаться его ощущение (Verity et al., 1982). У некоторых пациентов описана глухота (Verity et al., 1982). У большинства пациентов болезнь не выглядит прогрессирующей или же развивается очень медленно (Ferriere et al., 1992).
Некоторые случаи могут иметь более быстрое течение с клиническими признаками прогрессии (Johnson и Spalding, 1964), что согласуется с дегенерацией и регенерацией нервных волокон, выявляемых при биопсии нерва. Моторные скорости проведения сохранны или только немного уменьшаются, но сенсорные потенциалы действия не выявляются. Кроме того, у некоторых пациентов отсутствовали соматосенсорные вызванные потенциалы с нижних конечностей. Биопсия нерва часто показывает значительно атрофированные нервы. Количество миелинизированных аксонов сильно уменьшается, но немиелизированные волокна обычно в пределах нормы или незначительно уменьшены. Анализ связей двух многочисленных канадских семей позволил идентифицировать новый ген, HSN2, который располагается в пределах 8 интрона гена PRKWNK1 (Lafreniere et al., 2004). Функция HSN2 неизвестна. Мутация в том же самом гене была впоследствии идентифицирована в пораженной семье из Ливана.
в) НСВН III (семейная вегетативная дисфункция, синдром Райли-Дея). НСВН III является наиболее распространенной среди сенсорных и вегетативных невропатий. Заболевание преобладает среди евреев-ашкенази, у которых частота болезни между 0,5 и 1 на 10000 живых новорожденных с предполагаемой частотой носительства 1 на 50. Имеются редкие сообщения о случаях у нееврейских пациентов (например, Guzzetta et al., 1986). Заболевание передается как аутосомно-рецессивный признак, вызванный мутациями гена IKBKAP, который картирован на участке хромосомы 9q31 (Slaugenhaupt et al., 2005). Результаты гистопатологического исследования включают потерю нейронов в задних корешках, краевой зоне Лиссауэра и интермедиолатеральных столбах серого вещества (Pearson и Pytel, 1978) и потерю немиелинизированных и миелинизированных волокон в периферических нервах, где отмечается недостаток катехоламинэргических окончаний (Pearson et al., 1974).
Иммунологическая реактивность субстанции Р в substantia gelatinosa спинного мозга и в продолговатом мозге равным образом истощается (Pearson et al., 1982). Симпатические ганглии гипоплазированы.
Клинические проявления относятся главным образом к вегетативной нервной системе. Начало является врожденным, и гипотония, проблемы с сосанием, слабый крик и рвота присутствуют с рождения. Задержка роста становится заметной в дальнейшей жизни. Избыточное слезотечение не проявляется. Кожная сыпь, моторная дискоординация, нестабильность температуры и кровяного давления, циклическая рвота и слюнотечение варьируют. Типично относительное безразличие к боли (Axelrod et al., 1981). Восприятие температуры, потоотделение и иннервация кожи были изучены Hilz et al. (2004). Приступы апноэ и пневмония распространены, являясь обычно причиной смерти в грудном возрасте и детстве. Часто выявляются дилатация пищевода и нарушения моторики желудка. Постуральная гипотензия присутствует практически всегда. Большой проблемой является сколиоз. Глоточный рефлекс часто слабый. Диагностические критерии включают отсутствие грибовидных сосочков на языке, снижение или отсутствие глубоких сухожильных рефлексов, отсутствие слезотечения, миоз после закапывания в глаза 2,5% метахолина хлорида и отсутствие аксонального покраснения после гистаминовой инъекции внутри-кожно (Axelrod et al., 1974). Ни один из критериев не специфичен и может выявляться при других невропатиях. Пренатальный диагноз возможен.
Течение семейной вегетативной дисфункции тяжелое; ранние исследования показывают, что в 1960-е годы только 20% пациентов доживало до взрослого возраста, хотя к 1980-м за счет улучшения лечения показатель возрос до 50% (Axelrod и Abularrage, 1982). Осложнения со стороны пищеварительной и дыхательной систем распространены и могут усугубляться часто развивающимся кифосколиозом. Распространена эмоциональная неустойчивость с повторяющимися тяжелыми приступами задержки дыхания. Интеллект остается нормальным.
Лечение симптоматическое. Риск аспирационной пневмонии должен быть минимизирован за счет внимания к позе и тщательной профилактики во время кормления, что может потребовать питания через зонд, гастростомии или фундопликации. Диазепам эффективен в сочетании с хлорпромазином для лечения острых кризов и гипертензии. Лечение сколиоза затруднительно, в некоторых случаях возможна лишь частичная хирургическая коррекция искривления (Kaplan et al., 1997). Семьи больных детей нуждаются в значительной психологической поддержке.
г) НСВН IV (врожденная нечувствительность к боли с ангидрозом). При этом редком нарушении из-за мутации гена рецептора фактора роста нервов TRK/NGF отмечается отсутствие немиелинизированных нервных волокон в периферических нервах (Goebel et al., 1980). Также поражаются тракт Лиссауэра и дорсальные корешки спинного мозга. Заболевание возникает как врожденное с эпизодами необъяснимого повышения температуры, часто связанного с температурой окружающей среды. Типично отсутствие потоотделения (ангидроз). Нечувствительность к боли универсальна и приводит к ранам, самотравмированию и остеомиелиту, особенно нижних конечностей. Часто происходит прикусывание языка. Встречаются кожная сыпь и повышенная чувствительность зрачков к метахолина хлориду (Axelrod и Pearson, 1984). Как правило, имеется задержка интеллектуального развития, показатели IQ варьировали от 41 до 78, в большинстве случаев около 60 (Rosemberg et al., 1994). Моторные и сенсорные скорости проведения нерва нормальны или почти нормальны. Об умеренной форме без ангидроза сообщено Pavone et al., (1992).

Наследственная сенсорная невропатия IV типа.
Имеется очаговый дефицит тонких миелинизированных волокон.
Немиелинизированные волокна были очень разреженные при электронном микроскопическом исследовании.
д) НСВН V. НСВН V типа представляет собой врожденную нечувствительность к боли со сниженной тепловой чувствительностью, но с сохранением реакции на осязательные и механические стимулы и задержкой глубоких сухожильных рефлексов (Low et al., 1978). С патоморфологической точки зрения характерно практически полное исчезновение малых миелинизированных волокон и умеренным уменьшением числа немиелинизированных волокон. Результаты рутинных исследований моторного и сенсорного проведения по нерву в пределах нормы. Возможный локус для НСВН V на участке хромосомы 1р11.2-р13.2 был идентифицирован в многочисленной шведской семье со сниженным ощущением глубокой боли и температуры, но с нормальными когнитивными способностями. Анализ функционирующих генов-кандидатов в критической для болезни области показывает мутацию в кодирующем участке гена бета-рецептора фактора роста нерва (NGFb) (Einarsdottir et al., 2004).
е) Другие формы НСВН с нечувствительностью. Описано несколько редких и/или спорных типов НСМН. Дополнительные типы включают НСВН с дефицитом гормона роста (Liberfarb et al., 1993), прогрессирующую панневропатию с гипотензией (Axelrod и Pearson 1984), тип НСВН без трофических изменений (Bye et al., 1990), НСВН с нейротрофическим кератитом (Donaghy et al., 1987), врожденную сенсорную невропатию с ихтиозом и синдромом передней камеры глаза (Quinlivan et al., 1993), глухоту, сенсорную невропатию и овариальный агенез (Linssen et al., 1994), и НСВН с катарактами, олигофренией и поражениями кожи (Heckmann et al., 1995). НСВН, связанная со спастической параплегией, включает два различных типа, один поражает в основном мелкие сенсорные волокна (Cavanagh et al., 1979а), а другой — крупные волокна с немногочисленными невропатическими симптомами (Schady и Smith 1994). Кроме того, два различных типа сенсорной невропатии были описаны у детей индейского племени навахо (Appenzeller et al., 1976; Johnsen etal., 1993).
Некоторые случаи сенсорной невропатии могут имитировать жестокое обращение с детьми (Makari et al., 1994).
Термин нечувствительность к боли в принципе относится к пациентам, у которых аналгезия — результат патологии периферических нервов, нервных окончаний в коже или центральных сенсорных проводящих путей, тогда как безразличие к боли относится к тем, кто имеет нормальные сенсорные проводящие пути, но не в состоянии определить болезненную природу стимулов (Manfredi et al., 1981). Такое различие может быть во многом искусственным, и Dyck et al. (1983) подчеркнули факт, что точный анализ безразличия к боли с помощью современных методов показывает патологию периферической сенсорной системы. Однако сообщалось о случае с нормальным результатом морфометрического исследования нерва (Landrieu et al., 1990), и недавно описаны семейные случаи из-за мутаций в гене натриевого канала Navi.7 (Сох et al., 2006, Goldberg et al., 2007).
е) Смешанные сенсорные и вегетативные невропатии. Для редких случаев поражения вегетативной нервной системы характерна боль. Описан семейный доминантный синдром раннего начала пароксизмальной ректальной боли, связанной с одно- или двусторонней вазодилятацией в нижних конечностях и органах брюшной полости и часто вызываемой дефекацией. Позже возможна боль глазной и субмаксиллярной локализации. Во время приступов часто встречаются обмороки. Данное состояние, именуемое теперь пароксизмальным расстройством с чрезвычайной болью было определено как результат мутаций в гене рецептора Nav1.7 (Fertleman et al., 2007). Этот же ген мутирует и при эритромелалгии, характеризующейся приступами дистальной боли и эритемы, провоцируемой высокой температурой. Часто эффективно лечение карбамазепином.
Синдром беспокойных ног, который является особым типом сенсорной невропатии, частой у взрослых, также существует у детей (Kotagal и Silber, 2004). Для родственного с ним синдрома периодических движений конечностей во сне в 20% случаев характерно начало до 10-летнего возраста.
- Вернуться в оглавление раздела "Неврология."
Редактор: Искандер Милевски. Дата публикации: 11.1.2019
Рубрика МКБ-10: G60.8
Содержание
- 1 Определение и общие сведения
- 2 Этиология и патогенез
- 3 Клинические проявления
- 4 Другие наследственные и идиопатические невропатии: Диагностика
- 5 Дифференциальный диагноз
- 6 Другие наследственные и идиопатические невропатии: Лечение
- 7 Профилактика
- 8 Прочее
- 9 Источники (ссылки)
- 10 Дополнительная литература (рекомендуемая)
- 11 Действующие вещества
Наследственная сенсорно-вегетативная невропатия 4-го типа
Синонимы: врожденная нечувствительность к боли с ангидрозом
Наследственная сенсорно-вегетативная невропатия 4-го типа является наследственным расстройством, характеризующимся ангидрозом, нечувствительностью к боли, саморазрушительным поведением и эпизодами лихорадки.
Ген NTRK1 (TRKA), локализуется на хромосоме 1q21-22.
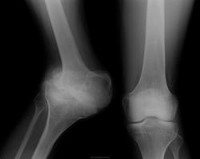
Наследственная сенсорно-вегетативная невропатия 4-го типа манифестирует в раннем детстве. Блихкородственные браки прослеживаются у 50% пациентов. Эпизоды лихорадки, выраженная гиперпирексия и рецидивирующие фебрильные судороги вследствии ангидроза, а также самоповреждения - являются самыми ранними симтомами расстройства. Кардинальной особенностью патологии является отсутствие или заметно сниженное потоотделение. Ангидроз отмечается на туловище и верхних конечностях в 100% случаев, другие области тела вовлекаются с разлтчной частотой. Кожа становится толстой, мозолистой, с лихенификацией ладоней, зонами гипотрихоза на голове и дистрофическими ногтями. Восприятие боли и температуры отсутствует. Со временем сенсорная нечувствительность возрастает, что приводит к самоповреждениям, ауто-ампутациям и рубцовым повреждениям роговицы. Пациенты имеют определенные проблемы в заживлении эктодермальных производных, переломы заживают медленно, крупные суставы подвержены повторяющейся травматизации и часто наблюдаются суставы Шарко и остеомиелит. Гипотония и задержка развития часты в первые годы жизни пациентов, но нормализуются с возрастом. Может присутствовать постуральная гипотензия с компенсаторной тахикардией. У менее 10% пациентов снижены глубокие сухожильные рефлексы. Чувствительность к вибрации нормальна или умеренно уменьшена. Сколиоз может присутствовать у 20% больных. Часто встречаются раздражительность, гиперактивность и склонность к гневу. Речь обычно понятна, однако могут возникнуть серьезные трудности с обучением.
Диагноз наследственной сенсорно-вегетативной невропатии 4-го типа требует наличия трех клинических критериев: ангидроза, снижения восприятия боли и интеллектуального дефицита, диагноз подтверждается фармакологическими тестами и невропатологическими данными. Из-за разнообразия мутаций ДНК-диагносьтка обычно не используется для подтверждения клинического диагноза. Морфология биопсии кожи обнаруживает дефицит волокон C и A-дельта в эпидермисе и отсутствующие или гипопластичные потовые железы без иннервации.
Дифференциальный диагноз включает другие наследственные сенсорно-вегетативные невропатии.
Лечение поддерживающее, ориентировано на борьбу с гипертермией, профилактику самоистязания и коррекцию ортопедических проблем, которые потенциально могут вызвать серьезные инвалидизирующие деформации.
Прогноз независимого функционирования пациентов зависит от степени выраженности заболевания и способности контролировать вторичные осложнения патологии.
Наследственная сенсорно-вегетативная невропатия 2-го типа
Определение и общие сведения
Наследственная сенсорно-вегетативная невропатия 2-го типа является наследственным расстройством, характеризующимся глубокой и диффузной потерей чувствительности с участием крупных и мелких нервных волокон и выраженной гипотонией.
Точная распространенность неизвестна, но оценивается как очень низкая, менее 50 случаев патологии описано.
Этиология и патогенез
Локус расположен в области 12p13.33. Наследственная сенсорно-вегетативная невропатия 2-го типа возникает спорадически передается как аутосомно-рецессивный признак.
Клинические проявления
Наследственная сенсорно-вегетативная невропатия 2-го типа проявляется в младенчестве или раннем детстве и не прогрессирует. Начальные симптомы, в период от рождения до 3 лет, включают серьезные проблемы с глотанием и кормлением, частые апноэ, самоповреждения и задержка развития. Часто наблюдается гастроэзофагеальный рефлюкс. Сенсорная дисфункция проявляется отсутствием восприятия боли, сильно сниженным восприятием температуры и сниженными глубокими сухожильными рефлексами (без мышечной атрофии или мышечной слабости). Ощущение позы, вкусовое восприятие, глоточный и роговичный рефлексы могут быть снижены. Чувство вибрации может быть нормальным. Трофические изменения присутствуют в верхних и нижних конечностях. Наследственная сенсорно-вегетативная невропатия 2-го типа сопровождается частым появлением травм, переломов конечностей, а также суставов Шарко. Потеря слуха наблюдается примерно у 30% пациентов, а постуральная гипотензия примерно у 25% пациентов. Зрачки имеют преувеличенный ответ на парасимпатомиметические средства, слезотечение происходит с задержкой. Периодически возникает гипергидроз или области ангидроза. Отсутствие аксонной вспышки после внутрикожного введения гистамина и отсутствие грибковых сосочков на языке являются характерными чертами патологии.

Врожденная сенсорная нейропатия с ангидрозом – крайне редкое наследственное заболевание, которое характеризуется нарушениями периферической иннервации и процессов потоотделения. Одними из главных симптомов этого состояния являются полное отсутствие ноцицептивной чувствительности, повышенная температура тела из-за нарушенной терморегуляции, приступы внезапной одышки. Диагностика заболевания основывается на данных физикального осмотра, проверки реакции организма на различные внешние раздражители, генетических исследований. Лечения врожденной сенсорной нейропатии с ангидрозом не существует, но больные требуют особого ухода.
- Причины врожденной сенсорной нейропатии с ангидрозом
- Симптомы врожденной сенсорной нейропатии с ангидрозом
- Диагностика врожденной сенсорной нейропатии с ангидрозом
- Лечение и прогноз врожденной сенсорной нейропатии с ангидрозом
- Цены на лечение
Общие сведения
Врожденная сенсорная нейропатия с ангидрозом (семейная дизавтономия типа 2, врожденная нечувствительность к боли с ангидрозом Свенсона) – очень редкая генетическая патология, обусловленная нарушениями иннервации и ноцицептивной чувствительности. Первые сообщения об этом состоянии появились в 1932 году, было обнаружено несколько семей из Пакистана, члены которых обладали полной нечувствительностью к боли, склонностью к самоповреждениям и повышенной температурой тела. На сегодняшний день определить встречаемость этого заболевания по причине его большой редкости не представляется возможным, всего описано около сотни больных с врожденной сенсорной нейропатией с ангидрозом.
Механизм наследования данного состояния аутосомно-рецессивный, мужчины и женщины поражаются с одинаковой долей вероятности. Для современной генетики и медицины в целом семейная дизавтономия типа 2 представляет огромный интерес по причине раскрытия генетических аспектов боли и возможности на этой основе создавать обезболивающие средства нового поколения.
Причины врожденной сенсорной нейропатии с ангидрозом
Выявлено два основных гена, мутации в которых ответственны за развитие данной патологии. Первый из них, NTRK1 расположен на 1-й хромосоме и кодирует особый рецептор нервных тканей (рецептор тирозинкиназы 1-го типа). Этот протеин является чувствительным к фактору роста нервов и при нарушении своей структуры, обусловленной мутацией гена NTRK1, не способен полноценно выполнять свои функции. В результате этого нарушается развитие холинергических нейронов, клеток симпатической нервной системы, чувствительных путей в задних корешках спинного мозга. Также затрудняются процессы миелинизации периферических волокон. Все это приводит к тому, что становится невозможным холинергическая иннервация потовых желез и проведение болевых нервных импульсов при сохранении тактильной, температурной и вкусовой чувствительности. Механизм наследования мутаций гена NTRK1 – аутосомно-рецессивный, по некоторым данным именно его дефекты обуславливают большую часть случаев врожденной сенсорной нейропатии с ангидрозом.
Вторым геном, мутации которого ассоциированы с этим заболеванием, является SCN9A, локализованный на 2-й хромосоме. Продуктом его экспрессии является особый белок (Nav1.7), относящийся к группе натриевых каналов нейронов. Выявлено, что на мембранах нейронов ноцицептивной системы наблюдается наибольшее количество именно таких каналов, но их конкретные функции там неизвестны. Заболевание, обусловленное мутацией гена SCN9A, также характеризуется полной потерей болевой чувствительности, однако другие симптомы, такие как повышение температуры, умственная отсталость выражены намного слабее. Поэтому клинически течение такой формы врожденной сенсорной нейропатии с ангидрозом является более благоприятным.
Симптомы врожденной сенсорной нейропатии с ангидрозом
Ведущим симптомом этого заболевания является полное отсутствие ноцицептивной чувствительности – больные с самого рождения не ощущают боли и не могут даже представить себе это ощущение. Они могут чувствовать прикосновение, холод, тепло – но болевых ощущений нет даже в тех случаях, когда на коже уже развивается ожог. Переломы костей, травмирование кожи и других частей тела могут ощущаться, но больные не описывают свои чувства как неприятные. В результате этого в детстве у них нередко наблюдается аутоагрессия – особенно часто пациенты кусают губы, язык, могут наносить себе другие повреждения. Это может сохраняться и во взрослом возрасте, особенно если врожденная сенсорная нейропатия с ангидрозом сопровождается умственной отсталостью – это характерно для состояний, обусловленных мутациями гена NTRK1.
У больных постоянно наблюдается повышенная температура тела, иногда с эпизодами лихорадки до 40-41°С, которая не реагирует на прием традиционных жаропонижающих средств. Это обусловлено нарушениями потоотделения, в результате чего не происходит обычного охлаждения тела. По этой же причине наблюдаются и изменения кожных покровов – на ощупь они горячие и сухие, сама кожа тонкая, часто легко собирается в складки. Среди других, менее постоянных симптомов врожденной сенсорной нейропатии с ангидрозом, выделяют раннее выпадение зубов, повышенный риск развития остеомиелита, некрозы костей и суставов. Причина этих изменений лежит в нарушениях иннервации ряда структур, немаловажную роль играет и позднее обращение больных к специалисту – одним из симптомов остеомиелита или некроза кости является боль, которую пациенты не способны ощутить. В редких случаях могут выявляться офтальмологические нарушения – кератоконус, наличие эрозий и язв роговицы.
Диагностика врожденной сенсорной нейропатии с ангидрозом
Для определения этого состояния применяют множество методик – физикальный осмотр, диагностические тесты для выявления ангидроза, генетические исследования, изучение наследственного анамнеза. При осмотре больных часто выявляются признаки аутоагрессии – сильно поврежденные губы и язык со следами от зубов (вплоть до отсутствия его части), могут быть свежие и старые порезы на теле и конечностях, признаки заживших переломов. Однако повреждения скелета или мышц могут быть обусловлены и тем, что при физической нагрузке больные не способны правильно оценить усилие и при этом рвут мышцы, связки или ломают кости. Температура тела повышена, кожные покровы сухие, может наблюдаться умственная отсталость различной степени. Больные чувствуют прикосновения, имеют нормальную температурную чувствительность, но уколы иголкой не воспринимают как боль, а скорее оценивают их как вариант осязания.
При проведении диагностических тестов не отмечается потовых реакций ни на какие раздражители – электростимуляция или пилокарпиновая проба полностью отрицательные. При повышении температуры окружающей среды потоотделение также не происходит, но начинает подниматься температура тела больного из-за затрудненной теплоотдачи. Электромиография не обнаруживает нарушений проводимости, все рефлексы полностью сохранены за исключением корнеального – он резко снижен или даже отсутствует, это является одним из важных признаков врожденной сенсорной нейропатии с ангидрозом.
Изучение наследственного анамнеза часто подтверждает семейный характер заболевания, генетическая диагностика сводится к прямому секвенированию гена NTRK1 с целью выявления мутаций. Некоторые лаборатории способны производить аналогичную процедуру и в отношении гена SCN9A. Врач-генетик может также производить поиск патологического гена у фенотипически здоровых носителей (в случае наличия врожденной сенсорной нейропатии с ангидрозом у родственников) или осуществлять пренатальную диагностику посредством амниоцентеза или биопсии ворсин хориона.
Лечение и прогноз врожденной сенсорной нейропатии с ангидрозом
Специфического лечения врожденной сенсорной нейропатии с ангидрозом не существует. С раннего детства необходим особый уход за больными – пресекать самоповреждения (кусание губ, языка), тщательно следить за состоянием костно-мышечной системы, контролировать температуру тела. При развитии лихорадки или перегрева устранить гипертермию возможно только физическими методами – холодным душем, использованием пузырей со льдом, обтиранием холодной водой. Если имеются признаки олигофрении, то больной должен находиться под постоянным присмотром, в случае сохраненного интеллекта взрослый человек с таким заболеванием теоретически может сам контролировать свое заболевание. При правильном уходе и здоровом образе жизни прогноз относительно жизни больного благоприятный.
Читайте также:
