
Синдром марфана влияние на нервную систему

Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Он дал четкое описание патологии, благодаря чему синдром получил его имя.
Что это такое?
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью.
В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[2], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Классификация
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Причины возникновения
Это аутосомно-доминантное состояние, генетическое заболевание, передается от родителей к ребенку через гены.
Вызван мутациями в гене FBN1. Мутации FBN1 связаны с широким континуумом физических функций, от изолированных особенностей до тяжелой и быстро прогрессирующей формы у новорожденных.
Особенности расстройства чаще всего обнаруживаются в сердце, кровеносных сосудах, костях, суставах и глазах. Некоторые особенности – например, расширение аорты (расширение основного кровеносного сосуда, которое переносит кровь от сердца к остальной части тела) – может быть опасным для жизни. Также могут быть затронуты легкие, кожа, нервная система. Патология не влияет на интеллект.
Распространенность, частота встречаемости составляет 1 из 5 000 человек, включая мужчин и женщин всех рас и этнических групп. Около 3 из 4 наследуют его, то есть получают генетическую мутацию от родителя, у которого она есть. Тип наследования при котором заболевший первый в семье- называется спонтанной мутацией. Существует 50-процентный шанс, рождения ребенка с генетической мутации от пораженных родителей.
Люди с синдромом Марфана рождаются вместе с ним, но особенности расстройства не всегда присутствуют сразу. У некоторых людей есть много особенностей при рождении, включая серьезные состояния, такие как расширение аорты. У других меньше симптомов, например, молодые не имеют признаков, пока не станут взрослыми. Некоторые особенности, особенно те, которые влияют на сердце и кровеносные сосуды, кости или суставы, со временем ухудшаются.
Это делает очень важным получение точной ранней диагностики и лечения. Без этого человек подвергается риску от потенциально опасных для жизни осложнений. Чем раньше началось лечение, тем лучше результаты.
Почти половина людей, страдающих синдромом Марфана, этого не знают.
Симптомы
Наиболее характерным признаком синдрома Марфана является сочетание поражений опорно-двигательной, зрительной и сердечно-сосудистой систем. Сроки их появления вариабельны, а проявления многообразны.
Обычно для постановки диагноза бывает достаточно присутствия следующих признаков:
- непропорционально длинные конечности;
- аневризма аорты;
- одно- или двухсторонняя эктопия хрусталика.
Однако кроме этих признаков существует еще около 30 других проявлений синдрома.

Одно- или двухсторонняя эктопия хрусталика при синдроме Марфана выявляется у 80 % пациентов. Обычно у таких людей развивается близорукость и астигматизм, но в некоторых случаях возникает и дальнозоркость. Чаще нарушения зрения происходят на 4-м году жизни ребенка. Далее они устойчиво прогрессируют.
Кроме этих заболеваний, при синдроме Марфана могут выявляться следующие патологии органов зрения:
- косоглазие;
- гипоплазия цилиарной мышцы;
- колобома радужной оболочки;
- увеличение размера и уплощение роговицы;
- изменение диаметра сосудов сетчатки;
- формирование катаракты;
- возникновение глаукомы в молодом возрасте.
У людей с синдромом Марфана могут выявляться следующие изменения в опорно-двигательной системе:
- рост намного выше среднего;
- астенический тип телосложения;
- длинный и узкий лицевой скелет;
- длинные конечности и пальцы;
- искривления позвоночного столба (спондилолистез, сколиоз, кифоз и др.);
- воронкообразная или килевидная деформация грудной клетки;
- маленький размер челюсти;
- аркоподобное высокое небо;
- чрезмерная гибкость и подвижность суставов;
- молоткообразная деформация пальцев стоп;
- протрузия вертлужной впадины;
- плоскостопие;
- патологии прикуса.
Средний рост людей с таким заболеванием при рождении может составлять у девочек 52,5 см (во взрослом возрасте около 175 см), у мальчиков 53 см (во взрослом возрасте около 191 см).
Из-за высокого неба и малых размеров челюсти у людей с синдромом Марфана могут возникать нарушения речи. Поражения скелета и суставных структур приводят к появлению артралгий и миалгий. Позднее такие изменения повышают риск развития раннего остеоартрита.
Доминирующими и наиболее опасными проявлениями синдрома Марфана становятся признаки поражений сердца и сосудов. Появляющиеся из-за повреждения структуры стенок сосудов эластического типа изменения (особенно аорты и легочной артерии) и пороки развития клапанов, перегородок сердца вызывают следующие симптомы:
- быстрое наступление усталости;
- учащенное сердцебиение;
- стенокардические боли с локализацией в спине, верхней конечности или плече;
- холодные руки и ноги;
- аритмии;
- одышка.
При выслушивании тонов сердца у таких пациентов могут определяться шумы, а при выполнении ЭКГ выявляются признаки стенокардии. При синдроме Марфана может развиваться кистозная медиальная дегенерация митрального или аортального клапана, приводящая к пролапсу этих клапанных структур. Кроме этого, у плода, унаследовавшего мутированные гены, с высокой долей вероятности могут развиваться врожденные пороки сердца. Иногда, при неблагоприятном течении такой неонатальной формы синдрома, у ребенка возникает прогрессирующая сердечная недостаточность, приводящая к летальному исходу до года жизни.
Однако наиболее характерным для синдрома Марфана поражением сердечно-сосудистой системы обычно становится прогрессирующее расширение, расслоение всходящей части аорты и появление на ней аневризм. Такие изменения провоцируются ослаблением соединительной ткани, приводящим к кистозной дегенерации сосудистой стенки. Быстрое прогрессирование таких поражений аорты может охватывать всю ее длину и ответвляющиеся от нее сосуды. Нередко такое осложненное течение патологии приводит к смертельному исходу.
Больные с синдромом Марфана не всегда имеют проблемы с легкими. Однако в некоторых случаях слабость соединительной ткани альвеол приводит к их удлинению и перерастяжению. Впоследствии у таких больных может возникать спонтанный пневмоторакс, эмфизема легких и дыхательная недостаточность. При отсутствии своевременной помощи и лечения такие патологии могут становиться причиной наступления летального исхода.
Кроме этого, у людей с синдромом Марфана может наблюдаться ночное апноэ, сопровождающееся прекращением дыхания во сне на 10 секунд и более.
У большинства детей уровень интеллекта соответствует норме и IQ составляет 85-115 единиц. У некоторых лиц с таким наследственным заболеванием уровень IQ существенно превышает верхние границы нормы.
Иногда у людей с синдромом Марфана могут присутствовать признаки неравномерной интеллектуальной деятельности и некоторые особенности личности, выражающиеся в завышенной самооценке, чрезмерной эмоциональности, плаксивости и раздражительности.
Одним из последствий синдрома Марфана может становиться дуральная эктазия, вызывающаяся растяжением и вытяжением соединительной ткани (оболочки), обволакивающей спинной мозг. Впоследствии эта патология может приводить к появлению болей и дискомфортных ощущений в брюшной полости или к слабости и неподвижности нижних конечностей.
Кроме вышеописанных проявлений синдрома Марфана в некоторых случаях могут выявляться следующие изменения в других органах и системах:
- атрофические стрии на коже;
часто рецидивирующие бедренные и паховые грыжи; - предрасположенность к растяжению и разрывам связок, подвывихам и вывихам;
- аномальное расположение почек;
- варикозное расширение сосудов;
- опущение матки и мочевого пузыря.
Большинство детей с синдромом Марфана тяжело переносят физическую нагрузку и после нее нередко ощущают боли в мышцах. Мышцы у таких больных могут быть недоразвитыми. На фоне психоэмоционального перенапряжения у детей могут периодически возникать приступы мигренеподобной головной боли. Кроме этого, нередко ощущается слабость и признаки гипотонии.
Диагностика
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Как лечить синдром Марфана
Лечение синдрома Марфана всегда требует одновременного участия нескольких специалистов: кардиолога, кардиохирурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лечебных процедур охватывает консервативные и оперативные способы лечения. Консервативные меры направлены на профилактику осложнений и поддержание нормального функционирования органов и систем, а оперативные предполагают коррекцию имеющихся анатомических изменений с целью предотвратить выраженное нарушение функций или даже угрозу для жизни больного.
Хирургические методы лечения:
- реконструктивные операции на аорте (при значительном, более 5 см, расширении восходящей части аорты и расслоении ее стенки);
- протезирование клапанов сердца;
- удаление измененного хрусталика с заменой его на искусственный;
- пластика позвоночника при выраженном сколиозе;
- эндопротезирование тазобедренных суставов;
- пластика грудной клетки (в последние годы отрицается ее целесообразность).
Больному необходим подбор очков или контактных линз, иногда возможна лазерная коррекция зрения.
Медикаментозное лечение имеет патогенетическую и симптоматическую направленность. Больному с метаболической целью назначают большие дозы витамина С (1-3 г в сутки), препараты с глюкозаминосульфатами и хондроитинсульфатами (Терафлекс, Структум, Хондроксид, Глюкозамин, Аминоартрин, Эльбона, Юниум), Карнитина хлорид 20% раствор, Янтарную кислоту по 100-200 мг 2 раза в день, препараты магния (например, Магне В6), поливитаминно-минеральные комплексы (с кальцием, магнием, цинком, медью). Прием этих веществ направлен на нормализацию обмена веществ, укрепление соединительной ткани.
Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
В целом подбор метода лечения и ассортимент применяемых лекарственных средств очень индивидуальны. Все зависит от спектра клинических симптомов и выраженности нарушений у конкретного больного.
Больным с синдромом Марфана показана лечебная физкультура, но в строго дозированном количестве, чтобы занятия приносили пользу сердечно-сосудистой и опорно-двигательной системам, а не вред. Нельзя заниматься контактными и игровыми видами спорта (баскетбол, футбол), рекомендовано плавание.

Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Исследуем симптомы и возможные осложнения, которые могут возникнуть в различных органах из-за такого генетического заболевания, как синдром Марфана.
Давайте посмотрим, как осуществляется диагностика и каковы на сегодняшний день наиболее эффективные методы лечения и увеличения продолжительность жизни.
Что такое синдром Марфана
Синдром Марфана – совокупность симптомов и признаков, составляющих клиническую картину серьезного генетического наследственного заболевания, которое поражает связочные ткани. Точнее, синдром Марфана является следствием мутации гена FBN1, расположенного на хромосоме 15, которая кодирует фибриллин-1, гликопротеин (белок, который содержит полимер, простые сахара или олигосахарид), они имеют важное значение для эластичности волокон соединительной ткани.
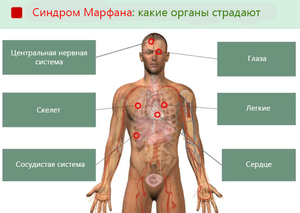
Таким образом, синдром Марфана влияет на множество важных органов и тканей: сердце, легкие, скелет, глаза и т.д.
Заболевание передается, как аутосомно-доминантный признак. Большинство больных наследует ген от одного родителей. Замечено, что эти случайные мутации имеют наиболее высокую вероятность проявления у пожилых родителей.
Соединительная ткань выполняет функцию поддержки, объединения и питания тканей организма, которые образуют различные органы. С точки зрения гистологии (наука, которая изучает состав тканей) соединительная ткань может быть различных типов, в зависимости от характеристик ткани и/или органа, которые она образует, но всегда характеризуется наличием клеток, разделенных между собой, но рассеянных в межклеточном веществе как внеклеточный матрикс.
Внеклеточный матрикс состоит из белков, которые образуют волокнистую часть фиброзной ткани и водный раствор белка.
Волокнистая ткань может состоять из трех различных типов волокон: коллагеновых, сетчатых и эластичных. Сетчатые и коллагеновые волокна имеют идентичный химический состав, но отличается структурной организацией. Эластические волокна состоят из двух отдельных белковых цепей в фибриллина и эластина.
Основными видами соединительной ткани являются:
Мы сказали, что Синдром Марфана является следствием мутации гена FBN1, который кодирует белок фибриллин-1. Он необходим для образования эластических волокон, которые составляют структуру соединительной ткани, а также содержат некоторые факторы роста, такие как TGF-beta.
Мутация FBN1 определяет снижение на физиологическом уровне фибриллина 1 и, следовательно, невозможность накопления TGF-beta и ухудшение волокон эластина, то есть возникновение синдрома Марфана.
Симптомы синдрома Марфана – зависят от органов
Учитывая большое количество органов, которые могут быть затронуты заболеванием, симптоматика может быть очень насыщенной (можно обнаружить более 30 различных симптомов и признаков). Кроме того, симптомы синдрома Марфана чрезвычайно изменчивы при переходе от одного лица к другому, даже если они члены одной семьи.

Синдром Марфана поражает.
У некоторых людей они имеют настолько легкую форму, что практически остаются незамеченными для окружающих, у других же симптомы имеют столь острую форму, что это ставит под угрозу жизнь человека.
Так же часто симптомы смешиваются с проявлениями других заболеваний. По этим причинам не всегда диагностика может быть проведена только на основании клинического анализа и в таких случаях будет необходимо прибегнуть к ДНК-тесту.
Возможные симптомы синдрома Марфана сгруппированы на основе влияния на функции различных органов.
- Рост выше нормы, сопровождается чрезмерной худобой.
- Сколиоз (искривление позвоночника).
- Кривые плечи.
- Аномальное искривление грудины.
- Длинные и тонкие верхние и нижние конечности (долихостеномелия), пальцы очень длинные и подвижные, напоминающие лапы паука (арахнодактилия).
- Плоскостопие.
- Очень глубокая вертлужная впадина (полость тазобедренного сустава, в которой расположена головка бедренной кости), что ограничивает движения головки бедренной кости.
- Высокое нёбо и стрельчатая арка, неправильный прикус. Из чего вытекают трудности с произношением звуков.
- Артроз, который развивается аномально рано.
Симптомы сердечно-сосудистой системы:
- Пролапс митрального клапана с астенией, сердцебиением и аритмией.
- Пролапс аортального клапана с недостаточностью аортального возврата крови в левый желудочек.
- Расширение восходящей и брюшной аорты с образованием аневризмы.
- Спонтанный пневмоторакс. Накопление воздуха в плевральной полости. В нормальных условиях давление на внешней поверхности легких меньше, чем атмосферное, и это приводит к тому, что легкие остаются раздутыми. Наличие воздуха в плевральной полости нарушает дыхательную функцию.
- Идиопатическое обструктивное заболевание легких. Болезнь, которая блокирует поток воздуха в легкие и затрудняет дыхание.
- Апноэ сна.
Симптомы центральной нервной системы:
- Дуральная эктазия. Выпячивание в мешочек, который покрывает и защищает спинной мозг. Проявляется болью в спине, головную болью, онемением и слабостью конечностей, невозможностью контроля сфинктеров, а затем недержанием мочи и кала.
- Дегенерация межпозвонковых дисков.
- Дисаутономия. Болезнь, которая влияет на нормальные функции вегетативной нервной системы и, следовательно, контроль частоты сердечных сокращений, кровяное давление и перистальтику (сокращение гладкой мускулатуры органов в виде трубки, таких как кишечник).
- Проблемы рефракции, такие как близорукость и астигматизм.
- Вывих хрусталика. Смещение хрусталика от его обычного положения.
- Отслоение сетчатки.
Диагностика синдрома Марфана: анамнез и показатели
Существует более 200 различных мутаций гена FBN1, молекулярные исследования позволяют определить более 95% случаев генетических дефектов. Но такие анализы могут занять много времени и довольно дорогие, поэтому почти всегда пытаются провести диагностику на основе клиники.
Учитывая большое количество симптомов и многие сопутствующие заболевания, чтобы облегчить задачу врачей, был разработан единый стандарт диагностики. Он предусматривает наблюдение за 7 аппаратами организма, а именно двигательным аппаратом, сердечно-сосудистой системой, легкими, глазами, нервной системой, генетическими маркерами, психикой.
Лечение синдрома Марфана – лекарства и вмешательства
Не существует никакого постоянного лечения синдрома Марфана, поэтому лечение, как правило, заключается в предотвращении и контроле возможных осложнений.
Лекарства при синдроме Марфана используются, в основном, чтобы сохранить под контролем артериальное давления для предотвращения чрезмерного увеличение аорты.
В последние годы используется метод лечения, действие которого основывается на ингибировании TGFβ. Данная терапия так же, главным образом, защищает от расширения аорты.
Также используют различные хирургические методы лечения для исправления некоторых недостатков, типичных для синдрома Марфана, такие как уменьшение сколиоза, исправление аорты, восстановление рефракции глаз.
Осложнения и риски синдрома Марфана

Синдром Марфана приводит к .
По мере того как органы будут затрагиваться в ходе развития синдрома Марфана, могут появиться различные осложнения, в частности:
- Аневризма и рассечение аорты, как восходящей, так и брюшной. Диссекция аорты часто отмечается во время беременности. В этом состоянии сердце матери подвергается чрезмерному напряжению, что может оказаться фатальным для целостности аорты.
- Нарушения митрального и/или аортального клапана. В результате того, что сердце „переутомляется“, что неизбежно ведет к сердечной недостаточности.
- Отслоение сетчатки.
- Вывих хрусталика и его дислокация.
- Возникновение ранней катаракты и глаукомы.
- Деформация скелета (грудина, позвоночник, ноги).
В прежние годы больные с синдромом Марфана редко жили дольше 40 лет. В настоящее время, с развитием методов хирургического вмешательства, пациенты в состоянии спокойно прожить до 70 лет.
Конечно, требуется постоянный мониторинг состояния организма, который позволяет осуществить своевременное вмешательство в случае критических ситуаций.
Наиболее частой причиной смерти является резекция аорты.

Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков. Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.

мальчик с синдромом Марфана
Клиническая симптоматика синдрома весьма полиморфна. Это связано с наличием пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью.
Больные имеют диспропорциональные конечности, рост выше среднего, вытянутые пальцы, гипермобильные суставы, худощавое тело, готическое небо, неправильный прикус, глубоко расположенные глаза. Они страдают гигантизмом, миопией, эктопией хрусталика, изменением формы грудины, кифосколиозом. Клинические признаки синдрома обусловлены гиперрастяжимостью тканей.

Синдром Марфана — наследственная коллагенопатия, встречающаяся у 1 из 10000-20000 человек. Патология обнаруживается повсеместно, среди людей любой национальности, пола и места проживания – одинаково часто как среди жителей юга, так и севера. В семейных парах, где отцу больше 35 лет, чаще рождаются больные дети.
Диагностика патологии основывается на данных наследственного анамнеза, визуального осмотра, результатах дополнительных исследований. Пациенты с синдромом Марфана внешне похожи друг на друга. Специалисты по внешнему виду могут предположить наличие недуга. Лечение болезни медикаментозное и оперативное, заключающееся в устранении структурных и функциональных нарушений в сердечной мышце, зрительном анализаторе, костно-суставном аппарате. Оперативное вмешательство показано в тех случаях, когда лекарственное и физиотерапевтическое лечение не дает положительных результатов. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Этиологические факторы
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
![]()
аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;- выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.

Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Классификация
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.

Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:

Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,
![]()
- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии – мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца – эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.

У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
![]()
страбизм,- катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.

Поражение нервной системы:
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
Поражение бронхопульмональной системы:
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.

Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Методы диагностики

проявления синдрома Марфана в младшем возрасте
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Методы лечения

Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
При снижении остроты зрения пациентам с офтальмологическими заболеваниями прописывают очки для постоянного ношения или контактные линзы.
- оперативное вмешательство на сердце и аорте — протезирование аорты и клапанов сердца, реконструктивные и пластические операции,
- операции на глазах — коррекция близорукости лазером, замена хрусталика, устранение глаукомы,
- хирургическая коррекция скелета — пластика грудной клетки при ее воронкообразной деформации, стабилизация позвоночника, протезирование крупных суставов.
Людям с марфаноидным фенотипом стертой формы показано физиолечение и ЛФК.
Клинические рекомендации специалистов своим пациентам:
- ведение здорового образа жизни,
- ограничение тяжелого труда и физического перенапряжения,
- отказ от спортивных игр повышенной активности,
- регулярное посещение специалистов в области кардиологии, офтальмологии, ортопедии, неврологии,
- нахождение больных под постоянным наблюдением и контролем врачей,
- периодическое прохождение диагностического обследования.
Прогноз и профилактические мероприятия

Прогноз синдрома Марфана неоднозначный. Он зависит от состояния кардиоваскулярной системы, глаз и скелета больного. При наличии серьезных нарушений со стороны этих органов длительность жизни пациентов ограничивается 40-50 годами, и повышается риск внезапной смерти. С помощью своевременной кардиохирургической коррекции можно улучшить качество жизни больных и вернуть им трудоспособность.
Все пары, в семейном анамнезе которых имелись случаи наследственных заболеваний, должны перед беременностью посетить генетика и заранее сдать все необходимы анализы. Пренатальная диагностика — комплекс мероприятий, проводимых с целью выявления патологии на стадии внутриутробного развития. Она заключается в проведении УЗИ плода и биохимического скрининга материнских сывороточных маркеров. К ее инвазивным методам относятся: биопсия ворсин хориона, исследование околоплодных вод, пуповинной крови и клеток плаценты.
Всем больным необходимо вовремя санировать имеющиеся очаги хронической инфекции в организме — лечить кариес, тонзиллит, синусит с помощью антибиотиков. Это крайне важное мероприятие, поскольку у лиц с синдромом Марфана иммунитет ослаблен. Им следует закаляться, правильно питаться, оптимизировать режим дня, полноценно спать, подолгу гулять на свежем воздухе, бороться с вредными привычками, избегать конфликтных ситуаций и стрессов.
Видео: синдром Марфана в программе “Жить здорово!”
Читайте также:
