
Синдромы эпилептические в младенчестве
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ

Эпилептическая энцефалопатия у детей – прогрессивное нарушение познавательных функций под влиянием эпилептического процесса. Проявляется дефицитом когнитивных, поведенческих функций, неврологическими отклонениями. Диагностика комплексная, включает клиническое обследование невролога и психиатра, энцефалографию, томографию, ультразвуковую допплерографию головного мозга, психодиагностическое исследование памяти, мышления, интеллекта, ряд лабораторных тестов. Лечение проводится противоэпилептическими препаратами, ноотропами, сосудорасширяющими, успокоительными средствами.
МКБ-10

- Причины
- Патогенез
- Классификация
- Симптомы эпилептической энцефалопатии у детей
- Осложнения
- Диагностика
- Лечение эпилептической энцефалопатии у детей
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Причины
Эпилептическое органическое поражение головного мозга возникает как результат воздействия наследственных и анамнестических факторов. Причинами патологии являются:
- Генетические изменения. Отдельные формы болезни развиваются при мутациях генов, хромосомных аномалиях (например, при синдроме Ангельмана).
- Метаболические сдвиги. Врожденными нарушениями метаболизма, приводящими к ЭЭ, являются некетотическая гиперглицинемия, метилмалоновая ацидурия, дефицит ксантиндегидрогеназы, синдром Зеллвегера, пропионовая ацидемия, D-глицериновая ацидурия.
- Новообразования мозга. Некоторые опухоли обладают эпилептогенным потенциалом, вовлечены в процесс формирования приступов (например, гамартома гипоталамуса).
- Пороки развития ЦНС. ЭЭ возникает на фоне гемимегалэнцефалии, порэнцефалии, синдрома Экарди, врожденного отсутствия сосцевидных тел, церебральной дисгенезии, фокальных кортикальных дисплазий.
- Поражения головного мозга. Повреждения ЦНС пренатального, натального и постнатального периода – распространенная причина эпилептической энцефалопатии у детей.
К факторам риска развития ЭЭ у ребенка относятся патологические процессы при беременности (инфекции, интоксикации, травмы), осложненные роды, перенашивание и недонашивание плода, эпилепсия у ближайших родственников, травмы головы.
Патогенез
Прогрессирующее когнитивное и нейропсихологическое ухудшение при эпилептических энцефалопатиях у детей объясняется агрессивной иктальной и электрической эпилептогенной активностью в период созревания головного мозга. Ее интенсивность определяется стадией созревания мозга, возрастом дебюта. У новорожденных отклонения ЭЭГ представлены вспышкой-подавлением, у младенцев – гипсаритмией, у детей раннего возраста – генерализованными разрядами медленных волн.
Эпиактивность в левом полушарии отражается изменениями речевых функций – диагностируется афазия, аграфия, акалькулия, алексия, речевая диспраксия. Вовлечение в патологический процесс правого полушария проявляется агнозиями, монотонностью речи, бедностью жестикуляции, нарушениями артикуляции. Активность в орбитофрональных, цингулярных областях, срединных структурах формирует отклонения в поведении – аутистические, агрессивные проявления, мутизм. Патологические изменения эмоций, специфической памяти возникают при очагах в гиппокампальных структурах, амигдале.
Классификация
Эпилептические энцефалопатии у детей классифицируют по характеру течения и особенностям клинической картины. Выделяют два типа патологий – I и II.
Эпилептическая энцефалопатия I характеризуется прогрессирующими нарушениями речи, интеллекта, когнитивных и опорно-двигательных функций, отклонениями поведения, эмоций. Сопровождается эпилептическими приступами, развивается в рамках следующих синдромов:
- Синдром Драве. Тяжелая миоклоническая эпилепсия младенчества. Проявляется ранними инфантильными фебрильными клоническими судорогами, миоклоническими и комплексными фокальными приступами, атипичными абсансами.
- Ранняя миоклоническая энцефалопатия. Приступ сопровождается хаотичным миоклонусом, за ним следуют простые фокальные приступы и тонические эпилептические спазмы.
- Эпилепсия со статусом медленного сна. Характеризуется фокальными ночными моторными приступами, осложненными абсансами, атоническими, клоническими или генерализованными тонико-клоническими приступами.
- Гипоталамическая эпилепсия. Наблюдаются тонические, атонические, тонико-клонические абсансы, нарушения сознания, немотивированный смех, реже плач.
- Синдром Ландау-Клеффнера. Выявляется вербальная слуховая агнозия, афазии, мутизм.
- Синдром Леннокса-Гасто. Отмечаются атонические, тонические, атипичные абсансы. В половине случаев – бессудорожный эпилептический статус.
- Миоклонический статус при непрогрессирующих энцефалопатиях. Миоклонии хаотичны, в периоды абсансов становятся ритмичными, синхронными.
- Синдром Отахара. Преобладают тонические спазмы (одиночные, кластерами). В трети случаев возникают хаотичные фокальные моторные клонические приступы, гемиконвульсии.
- Синдром Веста. Характеризуется инфантильными спазмами (кластеры), сопровождающимися плачем.
При эпилептической энцефалопатии II определяются нарушения эмоционально-поведенческой, когнитивной сферы. Наблюдается быстрая утомляемость, агрессивность, сниженная работоспособность, трудности концентрации внимания. Эпилептические приступы отсутствуют.
Симптомы эпилептической энцефалопатии у детей
Клиническая картина у детей представлена спектром психических и поведенческих расстройств, нейропсихологическими нарушениями, свойственными органическим поражениям мозга при неврологических болезнях. Энцефалопатические признаки носят специфический характер, зависят от возраста больного ребенка. У детей первого года жизни наблюдается беспокойство, капризность, беспричинная смешливость или плаксивость. Реакции на изменение освещения, звука, положения бывают неадекватными. Дети часто срыгивают, неспособны сосать, плохо спят. Сердцебиение неравномерное, мышечный тонус повышен.
Дошкольники страдают нарушениями сна, интенсивными головными болями. Сохраняется высокий тонус мышц, развиваются обморочные состояния. Эмоционально-волевая сфера отличается неуверенностью, перепадами настроения, чертами гневливости. Дети быстро утомляются, имеют проблемы с запоминанием, трудности переключения внимания. Появляются первые признаки ригидности психической деятельности. В школьном возрасте полностью разворачивается симптоматика когнитивного дефицита. Определяется снижение функции запоминания, усвоения новой информации. Недостаточная гибкость мышления проявляется трудностями адаптации, интеллектуальным дефицитом. Сохраняются расстройства сознания, головные боли, головокружения. Формируется эпилептоидный тип личности с раздражительностью, рассеянностью, депрессивными состояниями.
Осложнения
Эпилептическая энцефалопатия у детей приводит к патологиям двигательной, физической, речевой и психической сферы. Прогрессирующие формы осложняются деменцией, олигофренией, гидроцефалией, детским церебральным параличом, психозами, аффективными расстройствами, психопатологическими изменениями личности. Дети не осваивают школьную программу, социально дезадаптированы, нуждаются в уходе со стороны. Ранняя диагностика и адекватное лечение снижают вероятность осложнений, пациенты способны обучаться в обычной школе, энцефалопатия завершается формированием минимальной мозговой дисфункции.
Диагностика
Диагностика эпилептической энцефалопатии у детей – трудоемкое комплексное обследование. Используются клинические, лабораторные и инструментальные методы:
- Осмотр врача-невролога. Специалист уточняет жалобы, собирает анамнез, оценивает наличие патологических и сформированность нормальных рефлексов, их симметричность, координацию двигательной активности.
- ЭЭГ, МРТ, УЗДГ. Инструментальные исследования позволяют детально изучить структуру, функционирование, кровоснабжение головного мозга, выявить эпиактивность.
- Беседа с врачом-психиатром. В ходе опроса специалист определяет наличие органического симптомокомплекса, характер и выраженность поведенческих, эмоциональных, когнитивных отклонений.
- Психологическое тестирование. Исследование когнитивной, эмоционально-личностной сферы показано детям с 4-5-летнего возраста. Психолог использует патопсихологические пробы, проективные методики, опросники.
- Лабораторные тесты. Анализы выполняются с целью установления причины ЭЭ, определения синдрома. Проводятся тесты на исследование метаболизма, выявление аутоантител.
Дифференциальная диагностика эпилептических энцефалопатий основана на результатах инструментальных исследований мозга. Характерно наличие эпиактивности, ее связь с клиническими проявлениями, улучшение состояния при приеме антиконвульсантов (для большинства форм заболеваний).
Лечение эпилептической энцефалопатии у детей
Лечение подбирается индивидуально, зависит от множества факторов – возраста ребенка, степени тяжести эпилепсии, причин ее развития. Специфическая терапия базируется на применении медикаментов. Используются следующие группы препаратов:
- Противоэпилептические средства. Способствуют расслаблению мышц, устранению первичных и вторичных генерализованных судорожных припадков.
- Транквилизаторы. Подавляют возбудимость, эффективны при малых припадках.
- Ноотропы. Стимулируют активность нервных тканей, улучшают развитие памяти, внимания.
- Сосудорасширяющие препараты. Оптимизируют кровообращение, иннервацию в головном мозге.
- Аминокислоты, витамины. Стабилизируют обменные процессы в нервной ткани.
- Седативные средства. Применяются для коррекции эмоциональных нарушений.
Дополнением к медикаментозной терапии является психокоррекция когнитивных функций, логопедические занятия, массаж, лечебная физическая культура. Комплексный подход позволяет купировать основные симптомы эпилепсии, устранить осложнения (речевые, интеллектуальные, двигательные).
Прогноз и профилактика
Прогноз определяется формой заболевания, своевременностью диагностики и адекватностью терапии. Наиболее благоприятный исход отмечается при эпилептической энцефалопатии II, эпилепсии с непрерывными спайк-волнами медленного сна: происходит ремиссия приступов, когнитивные способности улучшаются, поведенческие и эмоциональные нарушения ослабевают, достигается приемлемый уровень социальной адаптации.
Профилактика эпилептической энцефалопатии осложнена, так как заболевание развивается очень рано. Снизить вероятность эпилепсии позволяет тщательная подготовка и ведение беременности: расчет рисков генетических отклонений, своевременное лечение заболеваний будущей матери, отказ от курения, употребления алкоголя.
Криптогенные и/или симптоматические
Таблица 3.8. Сравнительная характеристика инфантильных энцефалопатий
Обозначения: + наличие признака; — отсутствие признака.
Характеризуя ранние возрастзависимые эпилептические энцефалопатии, можно выделить несколько общих особенностей:
• жесткая зависимость от возраста; невозможность возникновения конкретной эпилептической энцефалопатий вне определенного возрастного диапазона;
• тяжелые и продолженные во времени эпилептические изменения на ЭЭГ;
• выраженная гетерогенность этиологии;
• частая ассоциация с моторными и ментальными нарушениями;
• весьма значительные затруднения в лечении и относительно неблагоприятный прогноз.
Такое значительное сходство указанных синдромов, а также использование нейроонтогенетического подхода при анализе ранних детских эпилепсий позволяют предположить, что именно ранний возраст, т.е. структурная и функциональная незрелость головного мозга, является основным фактором, детерминирующим развитие заболевания. Сами ранние эпилептические синдромы целесообразно рассматривать как неспецифическую реакцию незрелого мозга на экзо- или эндогенный стресс-фактор (возможно, один и тот же), реализующуюся эпилептическими припадками, тип которых зависит не от специфики фактора, а от степени структурно-функциональной состоятельности мозга.
Ранняя миоклоническая энцефалопатия. Ранняя миоклоническая энцефалопатия (РМЭ) — редкий возрастзависимый эпилептический синдром, впервые описанный J. Aicardi в 1978 г. В большинстве случаев заболевание начинается в возрасте, не превышающем 3 мес. Основным типом припадков являются миоклонии, преимущественно в виде фрагментарного мио-клонуса. Кроме того, могут наблюдаться частые внезапные парциальные приступы, массивные миоклонии и тонические спазмы.
Среди перечисленных клинических проявлений РМЭ патогномоничным признаком следует считать частые фрагментарные миоклонии, которые являются не только самым частым типом приступов, но и считаются дебютным, ранним симптомом заболевания. Тем не менее, с течением заболевания фрагментарные миоклонии постепенно уступают свою ведущую клиническую роль частым парциальным припадкам. Характеризуя особенности миоклонии, можно отметить, что они проявляются не только в состоянии бодрствования, но и во время сна. По степени выраженности они могут варьировать от легкого подергивания дистальных фаланг пальцев рук до миоклонии кистей, предплечий, век и угла рта. Частота их возникновения — от нескольких раз в день до нескольких десятков в минуту.
Парциальные припадки отмечаются приблизительно в 70—80 % случаев РМЭ и, подобно фрагментарным миоклониям, наблюдаются и во время бодрствования, и во время сна. Парциальные приступы при РМЭ отличаются большой частотой и могут достигать 300—500 раз в сутки.
К возрастному периоду 3—5 мес. супрессивно-взрывной ЭЭГ-паттерн постепенно замещается атипичной, или модифицированной, гипсаритмией, хотя в некоторых случаях он может персистировать достаточно долго.
Нейрорадиологические изменения при РМЭ не выражены. Как правило, и КТ, и МРТ не выявляют каких-либо грубых структурных изменений головного мозга. В тех редких случаях, когда они отмечаются, это преимущественно кортикальная атрофия различной степени выраженности.
Относительно этиологических аспектов РМЭ можно отметить, что не выявлено каких бы то ни было специфических этиологических факторов в развитии этого заболевания. Считается [Bernardina D., 1983], что определенную роль могут играть врожденные нарушения метаболизма (дизметабо-лические энцефалопатии раннего возраста), из которых выделяют как особо частые некетотическую гиперглицинемию, пропионовую ацидурию и D-глициновую ацидемию. В основном же большинство случаев РМЭ расценивается как криптогенные, т.е. формы, при которых презумптивно существующая причина, лежащая в основе развития эпилепсии, подразумевается, но не поддается идентификации на современном технологическом Уровне диагностических методов.
Лечение РМЭ составляет тяжелую и пока нерешенную проблему. К сожалению, к настоящему времени не существует антиконвульсантов и гормональных средств, которые могли бы обеспечить сколько-нибудь приемлемую эффективность лечения. Наиболее характерный исход заболевания смерть больных в первые 5 лет жизни; оставшиеся в живых страдают тяжелыми психомоторными расстройствами.
Ранняя эпилептическая энцефалопатия (синдром Отахары). Эта форма нцефалопатии является самым ранним по дебюту возрастзависимым эпилептическим синдромом. Она была впервые описана в 1978 г. японским ученым Shunsuke Ohtahara, а с 1989 г. признана в качестве самостоятельного эпилептического синдрома, получившего имя своего первооткрывателя — синдром Отахары.
В клинической картине приступы дебютируют в первые или 3 мес. жизни, но особенно часто в 1-й месяц. Основным типом припадков являются серийные или изолированные тонические спазмы. Приступы персистируют не только в состоянии бодрствования, но и ночью, помимо тонических спазмов, почти в половине случаев могут отмечаться моторные парциальные приступы, иногда по гемитипу. Миоклонические припадки нехарактерны, хотя в отдельных случаях могут иметь место.
Продолжительность тонического спазма при синдроме Отахары достигает приблизительно 10 с; в одной серии может отмечаться от 10 до 40 спазмов. Общее суточное количество спазмов достаточно велико и может достигать 300—400.
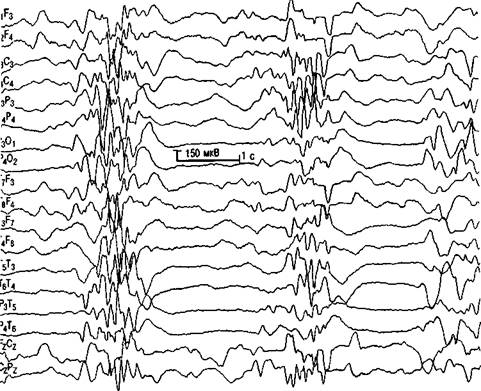
Основное ЭЭГ-проявление синдрома Отахары — описанный выше супрессивно-взрывной паттерн, который почти всегда отмечается и во сне, и в состоянии бодрствования. Вспышки медленных волн, длящиеся 1—3 с, имеют амплитуду 150—350 мкВ, перемежаются периодами почти полного уплощения ритма продолжительностью 3—4 с (рис. 3.9).

Рис. З.9. Ранняя инфантильная эпилептическая энцефалопатия (синдром Отахары). Диффузная супрессивно-взрывная активность.
В диагностике синдрома Отахары принципиально важны методы нейровизуализации, так как в отличие от РМЭ часто выявляют грубые структурные (порой асимметричные) изменения мозга. По данным S. Ohtahara (1997), эти нарушения отмечаются приблизительно в 85 % случаев ранней эпилептической энцефалопатии.
Как и РМЭ, синдром Отахары полиэтиологичен. В инициации заболевания особую роль играют врожденные мальформации головного мозга.
Мальформативные изменения мозга лежат в основе приблизительно 30—34 % случаев синдрома Веста [Jellinger К., 1987], по данным патоморфологических исследований. По своей классификационной сути это могут быть практически любые пороки развития мозга, включающие агенетические аномалии — агенезию мозолистого тела, агенетические порэнцефалические кисты, голопрозэнцефалию, агенезию червя и(или) гемисфер мозжечка; обширные эмбриоклас-тические процессы — гидранэнцефалию, эмбриокластические порэнцефалические кисты; гипо- и гиперпластические процессы — гипоплазию отдельных долей мозга, микроцефалию, унилатеральную мегалэнцефалию; кортикальные дисплазии — лиссэнцефалию, полимикрогирию, пахигирию, фокальную корковую дисплазию, ленточные и узловые нейронные гетеро-топии, микродисгенезии.
Особая роль в генезе инфантильных спазмов при синдроме Веста в последнее время уделяется микродисгенезиям, которые могут быть легко пропущены при рутинном диагностическом комплексе заболевания [Palm, 1986]. Предполагается, что небольшие участки дисплазированной ткани, Даже в небольшом количестве рассеянные в толще мозгового вещества, способны не только генерировать тяжелые и частые инфантильные спазмы, но и ответственны за грубые ментальные нарушения при синдроме Веста.
Ведущую роль в генезе инфантильных спазмов при этом заболевании безоговорочно отдают пери- и постнатальным гипоксически-ишемическим изменениям мозга [Chevrie, Aicardi, 1977].
Частота инфекционного фактора в развитии заболевания варьирует, по данным различных авторов, от 3 % [Matsumoto et , 1981] до И % [Lombroso C.T., 1983].
Инфекционные заболевания (цитомегалия, токсоплазмоз, герпетичес-й и краснушный энцефалиты, энтеровирусные и аденовирусные энцефалиты) могут играть более значимую роль в генезе инфантильных спазмов, так как вирусологические методы в настоящее время не позволяют проводить объективную оценку. Следует помнить, что свое катастрофическое эпилептогенное влияние они могут оказывать не напрямую, а опосредованно, например, через инициацию различных пороков развития.
Среди других (менее частых) причин возникновения синдрома Веста возмжно упомянуть нарушения метаболизма — до 10 % случаев [Meencke, srhard, 1985]; травматические изменения мозга (родовая травма); различие типы церебральных опухолей [Miyake et al., 1986].
Течение синдрома Веста в зависимости от этиологического фактора. В зависимости от этиологического фактора, лежащего в основе развития инфантильных спазмов при синдроме Веста, можно выделить несколько наиболее типичных типов течения заболевания:
• дальнейшее персистирование инфантильных спазмов в течение нескольких лет. Этот вариант течения заболевания чаще встречается при глубоких нарушениях кортикальной организации (особенно при диффузной лиссэнцефалии) или обширных эмбриокластических процессах (гидранэнцефалии);
• трансформация спазмов в мультифокальную эпилепсию. Как правило, такая процессуальность спазмов отмечается при множественных эпилептогенных нарушениях коры, например при туберозном склерозе или мультифокальной гипоксически-ишемической энцефалопатии. Результатом является появление мультифокальных или вторично-генерализованных приступов, относительно резистентных к антиком вульсантной терапии;
• трансформация спазмов в парциальную эпилепсию. Основные причины подобной трансформации включают порэнцефалию, очаговые формы кортикальных дисплазий и некоторые случаи туберозного склероза с единственным кортикальным туберсом;
• эволюция синдрома Веста в синдром Леннокса—Гасто. Это в основном отмечается при криптогенных инфантильных спазмах, т.е. спазмах, при которых этиологический фактор, лежащий в их основе, не идентифицируется, но подразумевается. Вообще отсутствие явных морфологических изменений мозга при столь губительной для прогноза трансформации инфантильных спазмов в синдром Леннокса— Гасто является одним из удивительных фактов эпилептологии, не поддающихся сколько-нибудь вразумительным объяснениям;
• полное спонтанное прекращение приступов. В подавляющем большинстве случаев отмечается у больных с криптогенными инфантильными спазмами.
Семиология эпилептических приступов при синдроме Веста.
Как уже говорилось, основным и единственным типом припадков при синдроме Веста являются инфантильные спазмы. Это один из кардинальных отличительных признаков заболевания от других ранних эпилептических энцефалопатий, в частности ранней миоклонической энцефалопатии. Спазмы представляют собой массивные генерализованные миоклонические или тонические сокращения аксиальной и конечностной мускулатуры.
Различают симметричные и асимметричные спазмы. При последних кинематика спазма имеет принципиально другой характер; наряду с типичными компонентами миоклонического или тонического инфантильного спазма отмечаются явные проявления парциального приступа, наиболее часто мимикрирующего асимметричный шейно-тонический рефлекс.
Продолжительность изолированного инфантильного спазма достаточно вариабельна и могут длиться от 0,5 до 2 сек.
Электроэнцефалографические аспекты синдрома Веста.
Основным (но не единственным) ЭЭГ-паттерном синдрома Веста является гипсаритмия. Гипсаритмия — это хаос и анархия на ЭЭГ.
Типичная гипсаритмия - это выраженное диффузное замедление активности, наличие диффузной быстрой активности; асимметрия фоновой записи.
При трансформации синдром Веста в синдром Леннокса—Гасто типичная функциональная гипсаритмия постепенно и неуклонно замещается модифицированной с преобладаем межполушарной синхронизации или генерализованной медленноволновой активности.

Рис. 3.10. ЭЭГ ребенка 5 мес с инфантильными спазмами. Модифицированная гипсаритмия.
Читайте также:
