
Число cag повторов при болезни гентингтона
| Число повторов | Риск развития заболевания у обследуемого и его детей |
| Менее 28 | Норма |
| 29-34 | Для обследуемого риска нет, но возможно увеличение числа повторов у детей и возникновение болезни |
| 35-39 | Возможно позднее развитие болезни |
| Более 40 | Болезнь |
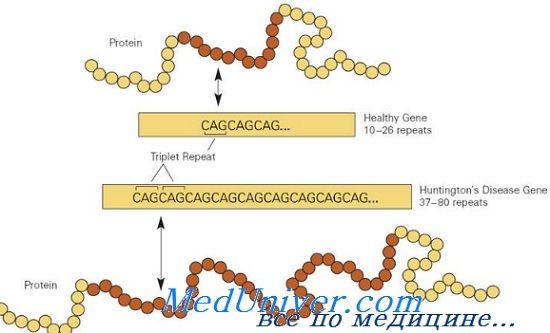
CAG-триплет кодирует глютамин. Поэтому в каждом из соответствующих этим генам белков имеется цепочка из последовательно расположенных глютаминов - полиглютаминовый трек. Болезнь развивается, когда длина этого полиглютаминового трека становится больше допустимой нормы. Оказалось, что удлиненные полиглютаминовые треки сами по себе или в составе полипептидных цепей способствуют агрегации белков в нейрональных клетках с образованием
нерастворимых комплексов. По мере накопления этих белковых агрегатов происходит гибель нейронов по типу апоптоза - програм- мированной гибели клеток. Этот процесс носит медленно прогрессирующий характер. Клинически нейродеградация проявляется тогда, когда погибает около 70% нейронов определенного типа. Дальнейшее развитие заболевания идет очень быстро, так как в оставшихся 30% нейронов также накапливаются нерастворимые белковые комплексы, что и приводит к их резкой деградации. Поэтому для всех нейродегенеративных заболеваний, обусловленных удлинением полиглютаминовых треков, характерны поздний дебют и тяжелое течение, достаточно быстро приводящие к летальному исходу. Заметим, кстати, что хорея Гентингтона может манифестировать не только в 40-50 лет, как это считалось ранее, а уже на 2-3-м десятилетии жизни, а в отдельных случаях намного раньше. Приводим следующее наблюдение, связанное с хореей Гентингтона. Пробанд В-а (родословная на рис. 26), 27 лет, обратилась в Лабораторию пренатальной диагностики наследственных заболеваний ИАГ им. Д.О. Отта РАМН (Санкт-Петербург) для уточнения диагноза хореи Гентингтона у своего мужа (Ш-2) 29 лет и прогноза состояния дочери (1У-1) 9 лет. У матери (11-3) отца девочки и ее бабушки (1-1) по отцовской линии наблюдается хорея Гентингтона. У мужа пробанда с 25 лет наблюдаются гиперкинезы дрожательного типа и отставание психического развития. При молекулярно-генетическом исследовании выяснилось, что муж пробанда от здорового отца получил 16 CAG-повторов, а от больной матери - 50. Эти данные четко подтверждают наличие хореи Гентингтона у мужа пробанда (см. табл. 1). Девочка (1У-1) унаследовала от отца нормальное число CAG-повторов - 16, но от матери она получила 33 CAG-повтора, т. е. премутацию. Последняя, к сожалению, не исключает развития заболевания у ее будущих детей, поэтому при вступлении в брак и беременности показана пренатальная диагностика хореи Гентингтона у плода. Отметим, что в детском возрасте хорея Гентингтона может протекать в виде эпилептиформноподобных пароксизмов, очень трудно поддающихся купированию, отставания психического развития и различного типа гиперкинезов. Таким образом, еще совсем недавно считавшаяся геронтологической патологией хорея Гентингтона благодаря ДНК-диагностике является и педиатрической проблемой. Как мы уже отмечали, заболевание диагностируют пренатально.
Увеличение числа CAG-триплетных повторов (экспансия) в 1 экзоне гена HTT является причиной развития болезни Гентингтона, аутосомно-доминантного неврологического заболевания.
Хорея Гентингтона, экспансия триплетных повторов, ген HTT, генетическое обследование.
Синонимы английские
Huntington disease, expansion of CAG (cytosine-adenine-guanine) triplet repeats, HTT gene.
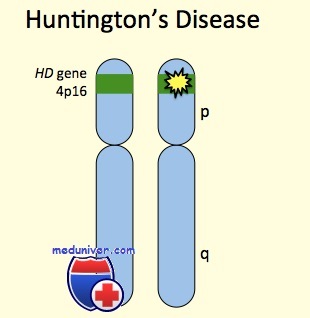
Локализация гена на хромосоме
Фрагментный анализ 1 экзона гена HTT.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Болезнь Гентингтона (БГ) – наследственное прогрессирующее нейродегенеративное заболевание, вызванное экспансией CAG-триплетных повторов в 1 экзоне гена HTT, располагающегося на коротком плече 4 хромосомы и кодирующего белок гентингтин (HTT). Небольшое количество нуклеотидов CAG (цитозин-аденин-гуанин) в гене HTT встречается в норме, однако при увеличении числа повторов (экспансии) более 35 вероятность развития заболевания и передачи гена потомкам значительно увеличивается. Размер экспансии коррелирует с тяжестью симптоматики и временем первых проявлений заболевания.
Встречаемость заболевания среди населения с европейскими корнями составляет 10:100000. Заболевание наследуется по аутосомно-доминантному типу, то есть имеется 50% риска его развития у потомков.
Болезнь Гентингтона проявляется преимущественно неврологической симптоматикой. Клинические проявления и признаки:
- Моторные нарушения - гиперкинезы: хорея, дистония, атетоз, тремор, миоклония; окуломоторные нарушения; брадикинезия и ригидность; дисфагия; нарушение равновесия, координации и походки.
- Когнитивные нарушения - снижение исполнительных повседневных функций; деменция.
- Нейропсихические нарушения - депрессия; раздражительность, агрессивность; психозы (бред, галлюцинации, паранойя, сверхценные идеи); апатия; персеверация и обсессивно-компульсивное расстройство.
- Инструментальное обследование - МРТ головного мозга: атрофия головки хвостатого ядра, проявляющаяся увеличением фронтальных рогов латеральных желудочков головного мозга. Уменьшение объема коры головного мозга. При МР-спектроскопии: увеличение лактата в области затылочной коры; уменьшение индекса NAA/creatine. При исследовании ПЭТ/КТ: гипометаболизм базальных ганглиев и фронтальной коры.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на болезнь Гентингтона проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- При подозрении на болезнь Гентингтона;
- при дифференциальной диагностике гиперкинезов;
- при дифференциальной диагностике хореи и дистонии;
- при когнитивных и нейропсихических нарушениях;
- при выявлении характерных изменений при проведении МРТ-исследования головного мозга;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на подсчете числа тройных CAG-повторов с помощью метода фрагментного анализа в гене НТТ. Диагностическая значимость обнаруженного числа CAG-повторов в гене HTТ представлена в таблице:
Количество CAG-повторов в гене HTT
Диагностический тест
Прогностический тест
Риск развития для детей
6-26 – нормальная аллель
Болезнь Гентингтона исключена
Болезнь Гентингтона не разовьется
Невысокий риск развития болезни Гентингтона
27-35 – умеренное увеличение
Болезнь Гентингтона очень маловероятна
Маловероятно, что болезнь Гентингтона разовьется
Риск развития БГ ( Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, риска прогрессирования заболевания и назначения лечения рекомендуется получить консультацию специалиста.
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
43 Генетическое обследование на гентингтоноподобное заболевание 2 типа в гене JPH3
55 Генетическое обследование на гентингтоноподобное заболевание 4 типа в гене TBP
43 Генетическое обследование на дентаторубро-паллидолюисову атрофию в гене ATN1
51 Комплексное обследование на гентингтоноподобные заболевания (ГПЗ2,ГПЗ4,ДРПЛА)
69 Медь в крови
105 Медь в моче
48 Обследования на частые генетические причины мозжечковой атаксии (СЦА 1,2,3,6,7, АФ)
42 Определение экспансии триплетов при спиноцеребеллярной атаксии 1 типа (в гене ATXN1)
43 Определение экспансии триплетов при спиноцеребеллярной атаксии 2 типа (в гене ATXN2)
42 Определение экспансии триплетов при спиноцеребеллярной атаксии 3 типа (в гене ATXN3)
45 Определение экспансии триплетов при спиноцеребеллярной атаксии 6 типа (в гене CACNA1A)
43 Определение экспансии триплетов при спиноцеребеллярной атаксии 7 типа (в гене ATXN7)
31 Диагностика системной красной волчанки
Литература
- Martha Nance et al., A Physician’s Guide to the Management of Huntington’s disease (Third Edition), HDSA 2011.
- Robert B. Daroff, Bradley's Neurology in Clinical Practice, 2-Volume Set, 7th Edition,Elsevier, 2016.
- Losekoot M, van Belzen MJ, Seneca S, Bauer P, Stenhouse SaR, Barton DE. EMQN/CMGS best practice guidelines for the molecular genetic testing of Huntington disease. Eur J Hum Ge net. 2013; 21(5): 480-486.
- Bean L, Bayrak-Toydemir P. American College of Medical Genetics and Genomics Standards and Guidelines for Clinical Genetics Laboratories, 2014 edition: technical standards and guidelines for Huntington disease. Genet Med. 2014; 16(12): e2.
Этиология и встречаемость болезни Гентингтона. Болезнь Гентингтона (MIM № 143100) — панэтническое аутосомно-доминантное прогрессирующее нейродегенеративное заболевание, вызванное мутациями в гене HD. Распространенность болезни Гентингтона колеблется от 3 до 7 на 100 000 среди западных европейцев до 0,1—0,38 на 100 000 среди японцев. Эти изменения распространенности отражают изменения в распределении аллелей болезни Гентингтона и гаплотипов, предрасполагающих к мутации.
Продукт гена HD, белок генгтинтин, экспрессируется повсеместно. Функция генгтинтина остается неизвестной.
Болезнетворные мутации в гене HD обычно вызваны экспансией последовательности повторов CAG в экзоне 1, кодирующем полиглутаминовую цепочку; в норме аллели HD имеют от 10 до 26 повторов триплета CAG, тогда как мутантные аллели имеют больше 36 повторов. Приблизительно у 3% пациентов болезнь Гентингтона развивается в результате новой экспансии повторов CAG; 97% наследуют мутантный аллель HD от больного родителя.
Новые мутантные гены болезни Гентингтона возникают вследствие перехода премутации (27-35 повторов CAG) в полную мутацию. До настоящего времени все описанные пациенты наследовали новую полную мутацию от отца.
Экспансия полиглутаминового участка генгтинтина приводит к появлению у него нового свойства, и как необходима, так и достаточна для индукции болезни Гентингтона. Кроме выраженной рассеянной атрофии неостриатума, основного признака болезни Гентингтона, экспрессия мутантного генгтинтина вызывает дисфункцию нейронов, общую атрофию мозга, изменения уровней нейромедиаторов и накопление ядерных и цитоплазматических агрегатов в нейронах. В итоге экспрессия мутантного генгтинтина вызывает гибель нейронов; тем не менее похоже, что клинические симптомы и дисфункция нейронов предшествуют развитию внутриклеточных агрегатов и смерти нейронов. Механизм того, как экспрессия увеличенного числа глутаминовых остатков вызывает болезнь Гентингтона, остается неясным.
Возраст начала болезни обратно пропорционален числу повторов CAG в гене HD. Пациенты с началом болезни во взрослом возрасте обычно имеют 40-55 повторов; в юношеском возрасте — обычно более 60 повторов. Пациенты с 36-41 повторами CAG имеют неполную пенетрантность, т.е. у них болезнь может не развиться в течение всей жизни. За исключением возраста начала, число повторов не влияет на другие проявления болезни Гентингтона.
Нестабильность и экспансия повторов CAG часто приводит к явлению антиципации, т.е. более раннему возрасту начала в последующих поколениях семьи. Достигнув однажды значения 36, число повторов увеличивается в ходе сперматогенеза отца; экспансия при материнской передаче мутации встречается реже. Поскольку число повторов триплета CAG обратно коррелировано с возрастом начала, индивидуумы, унаследовавшие мутацию от отца, имеют повышенный риск развития болезни с ранним началом; приблизительно 80% молодых пациентов наследуют мутантный ген болезни Гентингтона от отца.
Приблизительно треть пациентов имеют психиатрические нарушения; две трети — комбинацию познавательных и двигательных нарушений. Средний возраст пациентов на начало болезни — 35-44 года; приблизительно у четверти пациентов болезнь Гентингтона проявляется после 50 лет и у одной десятой — до 20 лет. Среднее выживание после установления диагноза — 15-18 лет, а средний возраст смерти — 54-55 лет.
Для болезни Гентингтона характерны прогрессирующие двигательные, познавательные и психиатрические расстройства. Двигательные нарушения включают как произвольные, так и непреднамеренные движения. Первоначально эти движения создают незначительные помехи ежедневной деятельности, но обычно приводят больного к нетрудоспособности по мере развития болезни.
Хорея, присутствующая у более чем 90% пациентов, — наиболее частая форма непроизвольного гиперкинеза; характеризуется неповторяющимися непериодическими толчками, которые не могут быть подавлены усилием воли. Познавательные нарушения начинаются в начале болезни и влияют на все аспекты познания; речь обычно затрагивается позднее, чем другие функции.
Поведенческие нарушения, обычно развивающиеся в ходе болезни, включают антиобщественное поведение, агрессию, взрывы гнева, апатию, сексуальные отклонения и повышенный аппетит. Психиатрические проявления, развивающиеся на любой стадии болезни, включают изменения личности, легкие психозы и шизофрению.
В конечных стадиях болезни Гентингтона обычно развиваются настолько выраженные двигательные нарушения, что больные полностью зависят от постороннего ухода. Они также теряют массу тела, у них появляются нарушения сна и мутизм. С течением болезни поведенческие нарушения уменьшаются.
Особенности фенотипических проявлений болезни Гентингтона:
• Возраст начала: от подросткового до старости
• Двигательные нарушения
• Познавательные нарушения
• Психиатрические нарушения
К настоящему времени никакого эффективного лечения при болезни Гентингтона нет. Помощь сводится к уходу и фармакологическому лечению поведенческих и неврологических проблем.
Каждый ребенок родителя с болезнью Гентингтона имеет 50% риск унаследовать мутантный аллель болезни Гентингтона. За исключением аллелей с неполной пенетрантностью (36-41 повтор CAG), у всех детей, унаследовавших мутантный аллель, если они имеют нормальную продолжительность жизни, развивается болезнь Гентингтона.
Дети отцов, несущих премутацию, имеют эмпирический приблизительно 3% риск унаследовать аллель болезни Гентингтона, в котором премутация перешла в полную мутацию. Тем не менее не все мужчины, несущие премутацию, равновероятно передают полную мутацию.
Доклиническое тестирование и пренатальная диагностика доступны благодаря анализу числа повторов CAG в 1 экзоне гена HD. Доклиническое тестирование и пренатальная диагностика — методики прогностические, поэтому лучше интерпретируются в случае подтверждения экспансии CAG у больного члена семьи.
Пример болезни Гентингтона. М.П., 44-летний мужчина, отметил ухудшение памяти и внимания. По мере снижения интеллектуальных функций в течение последующего года у него развились непроизвольные движения пальцев кистей и стоп, а также мимических мышц лица типа гримасничания. Он понимал свое состояние и впал в депрессию. До этого М.П. был здоров, и в его анамнезе не было аналогично больных родственников; его родители погибли в сорокалетнем возрасте в автокатастрофе. У пациента одна здоровая дочь.
После углубленного осмотра невролог диагностировал состояние больного как болезнь Гентингтона. Диагноз болезни Гентингтона подтвержден анализом ДНК, показавшим наличие 43 повторов триплета CAG в одном из аллелей гена HD (в норме не более 26). Последующее пресимптоматическое обследование дочери показало, что она также унаследовала мутантный аллель HD. Оба получили подробную консультацию.
Сегодня, по данным Гарвардского центра нейроисследований, только в США около 5 млн человек страдают от болезни Альцгеймера; 1 млн – от болезни Паркинсона; 400 тыс. – от рассеянного склероза; по 30 тыс. – от бокового амиотрофического склероза (болезнь Лу Герига) и болезни Гентингтона. Эти расстройства, относящиеся к группе нейродегенеративных заболеваний, плохо изучены, и для них отсутствуют эффективные способы лечения. Решить эту проблему может помочь инструмент редактирования генов – система CRISPR/Cas9, которая на сегодняшний день активно и успешно применяется в биологии и медицине
Рассмотрим эту проблему на примере болезни Гентингтона (другие названия – синдром Гентингтона, хорея Гентингтона или Хантингтона), проявляющейся такими симптомами, как нарастающая потеря двигательного контроля и психические расстройства. Несмотря на то, что генетическая мутация, вызывающая это заболевание, была выявлена более 20 лет назад, молекулярные механизмы его развития все еще до конца не выяснены, так же как не найдено эффективной терапии этой болезни.
Моделируем болезнь
Причина болезни Гентингтона – увеличение числа тринуклеотидных повторов CAG в гене НТТ, кодирующем белок гентингтин. В норме в этом гене содержится от 8 до 36 повторов, но, когда это число достигает 37 и более, начинается развитие заболевания. Триплет CAG кодирует аминокислоту глутамин. Соответственно, при болезни Гентингтона синтезируется гентингтин с удлиненной полиглутаминовой цепочкой. Такой белок имеет неправильную пространственную структуру и не может выполнять свою функцию в клетках, которая, кстати сказать, до сих пор точно неизвестна. Кроме того, в стриатуме, одном из отделов мозга, регулирующих мышечный тонус, мутантный белок формирует агрегаты, обладающие токсическим эффектом. При этом тяжесть протекания болезни напрямую зависит от числа повторов CAG.
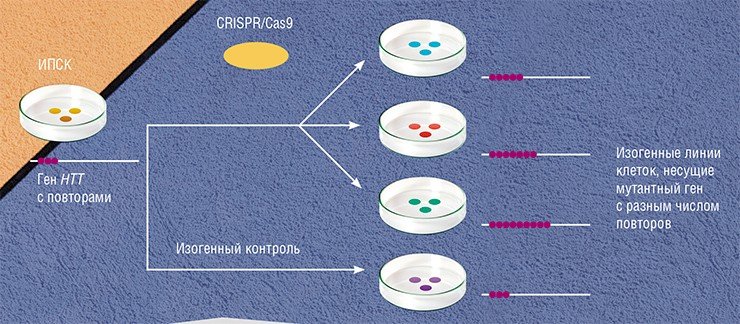
Для изучения болезни Гентингтона необходимо иметь клетки стриатума, но получить такой биоматериал затруднительно. Эта проблема решается с помощью индуцированных плюрипотентных стволовых клеток (ИПСК), которые достаточно легко с помощью перепрограммирования можно получить из клеток кожи или крови человека. Далее эти клетки можно наращивать в неограниченных количествах и дифференцировать (превращать) в практически любой нужный тип клеток.
Исследуем генное окружение
Несмотря на то, что болезнь Гентингтона является моногенным заболеванием, т. е. обусловлена мутацией в одном гене, известно более 100 так называемых генов-модификаторов, которые могут повлиять на время возникновения первых симптомов, тяжесть заболевания и т. д. Наиболее удобным инструментом для выяснения роли этих генов в патогенезе болезни Гентингтона является система GeCKO на основе CRISPR/Cas9 (Shalem et al., 2014). Она позволяет создавать целые библиотеки генных мишеней для CRISPR/Cas9 и выключать тысячи генов всего лишь за пару этапов. Таким образом, можно за короткий срок произвести нокаут (выключение) всех генов-модификаторов и выяснить, как это влияет на жизнеспособность клеток, скорость их деления и т. д.
Лечим?
CRISPR/Cas9 можно использовать и для лечения болезни Гентингтона. Есть два варианта воздействия: укорочение повторов CAG или специфическое выключение мутантной копии гена.
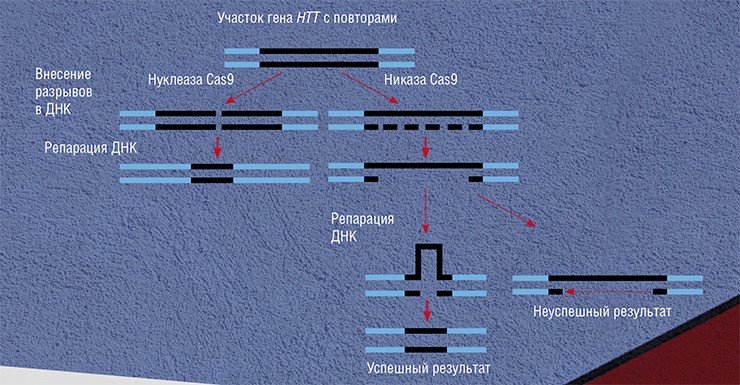
В первом случае с помощью CRISPR/Cas9 вносятся двунитевые разрывы в последовательность с повторами. Однако такое вмешательство имеет и обратный результат – увеличение числа повторов CAG. Проблема была решена путем использования никаз Cas9 – мутантных белков Cas9, которые вносят в ДНК однонитевые разрывы. Эксперимент показал, что последовательные однонитевые разрывы в регионах с повторами в худшем случае не изменяют длину участка с повторами, а в лучшем – приводят к его ожидаемому укорочению (Cinesi et al., 2016).
Исследования болезни Гентингтона с применением системы CRISPR/Cas9 сегодня проводятся и в России. Так, в лаборатории эпигенетики развития Института цитологии и генетики СО РАН (Новосибирск) ведутся работы по получению изогенных линий стволовых клеток, моделирующих болезнь Гентингтона, путем внесения удлиненных трактов CAG в нормальный ген НТТ, а также по дифференцировке ИПСК в нейроны стриатума. В дальнейшем новосибирские исследователи планируют перейти к изучению роли генов-модификаторов в патогенезе этой болезни с использованием клеточной модели.
Есть основания надеяться, что результаты исследований приведут к созданию эффективного метода лечения. Кроме того, они, возможно, помогут при изучении других нейродегенеративных заболеваний, развитие которых обусловлено подобной мутацией.
Медведев С. П. Как отредактировать наследственность // НАУКА из первых рук. 2014. Т. 55. № 1. С. 10—14.
Cinesi C., Aeschbach L., Yang B., Dion V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase // Nat. Commun. 2016. V. 7. P. 13272—13281.
Shalem O., Sanjana N. E., Hartenian E. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells // Science. 2014. V. 343. N. 6166. P. 84—87.
Shin J. W., Kim K.-H., Chao M. J., et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9 // Hum. Mol. Genet. 2016. V. 25. N. 20. P. 4566—4576.

Болезнь Гентингтона (код по МКБ-10 – G10) – это очень редкое наследственное нейродегенеративное заболевание, вызываемое нарушениями клеток в мозге (в стриатуме) с их последующим отмиранием и поражением ЦНС.
Описание
Хорея Гентингтона – что за болезнь, в чем ее суть? Это серьезное наследственное заболевание с типичными психическими и физическими симптомами, которые обычно возникают между 30 и 45 годами. Но признаки могут появиться раньше. Если они наблюдаются в возрасте около 20 лет, патология упоминается как ювенильная болезнь Гентингтона (вариант Вестфаля). Как правило, она характеризуется неконтролируемым подергиванием конечностей, туловища, головокружением. Заболевание приводит к психологическому расстройству личности, слабоумию.
Синдром Гентингтона затрагивает около 5-10 человек на 100000. К сожалению, ее невозможно предотвратить, замедлить или вылечить. Ожидаемая продолжительность жизни уменьшается не самой болезнью, а ослабленным иммунитетом.
Патология получила свое название в честь американского врача Джорджа Гентингтона, впервые описавшего ее в 1872 г.
Причины и факторы риска
Причина заболевания – изменение (мутация) гена IT15 на 4-й хромосоме.
Ген, содержащий менее 36 триплетов, продуцирует белок хантингтин, необходимый для правильного развития мозга. Болезнь Гентингтона – это заболевание с аутосомно-доминантным типом наследования. Это означает, что она встречается в основном в каждом поколении. Если один из родителей страдает этим расстройством, существует 50% риск того, что ребенок также заболеет.

При диагностике болезни Гентингтона необходимо учитывать, что не всегда известно присутствие в семье носителя мутаций. Причины – преждевременная смерть от другой патологии, развод, неизвестность отца. Новые мутации встречаются очень редко.
Пациентам, страдающим от заболевания, рекомендуется не иметь детей. Но проблема заключается в том, что в большинстве случаев оно диагностируется только после рождения ребенка. Генетическая мутация присутствует с рождения, но обычно начинает проявляться во взрослом возрасте.
Симптомы
Человек обычно приходит к врачу из-за проблем с движением, сопровождающихся изменением психики. Симптомы и признаки болезни Гентингтона чаще всего появляются около 40 лет. Клиническая картина постепенно ухудшается в результате повреждения головного мозга.
Вначале наблюдаются мелкие мышечные подергивания и постоянные движения конечностей. Позже развивается полная хорея. Она сначала поражает руки и голову, человек делает различные гримасы, открывает рот. Затем затрагиваются нижние конечности. Эти непроизвольные движения значительно усиливаются при стрессе, в то же время, вообще не появляются во время сна. Движения прогрессируют, человек становится неспособным к самостоятельности. Признаки проявляются на обеих сторонах тела.
Типичный симптом болезни хорея Гентингтона – нарушение походки, которая иногда напоминает танец. На более поздней стадии происходят падения, неуверенные движения. Непроизвольные движения могут больше не проявляться, развивается ригидность, человеку трудно двигаться.
Заболевание часто сопровождается нарушением речи (она становится непонятной). Кроме того, наблюдается расстройство глотания, приводящее к серьезным проблемам с приемом пищи. Пищевые расстройства вызывают потерю веса.
В большинстве случаев при болезни Гентингтона возникает недержание мочи.
Значимые проявления генетического заболевания хорея Гентингтона – психические проблемы. Первоначально возникает незначительная раздражительность, затем появляются другие признаки, такие как:
- агрессивность;
- апатия;
- безжалостное поведение;
- склонность к воровству;
- сниженный интерес к внешности;
- общие изменения личности.
Хотя окружающие замечают эти проблемы, немногие приписывают их болезни.
Депрессия возникает уже на ранних стадиях заболевания. Появляется страх перед новым днем, опасения потери производительности, неудач, нехватка энергии, воли к действию. Человек становится безразличным к окружающему миру, раздражается, имеет трудности с чувством радости, не концентрируется, страдает нарушением памяти. В результате психических проблем возникает снижение аппетита, потеря веса, нарушения сна. Больного могут даже посещать мысли о самоубийстве. Частью депрессии бывает тревожное расстройство.
Депрессия может чередоваться с периодом гипомании, которому присуще чувство полноты энергии, активности.

Больному не хватает проницательности и самокритики, поэтому ему трудно понять, что он больше не может продолжать принимать решения о содержании семьи или управлять автомобилем.
Серьезные проблемы включают галлюцинации и бред. Галлюцинация – это расстройство восприятия, при котором пациент не может провести различие между реальностью и картинами, которые рисует его воображение. Он воспринимает звуковые и визуальные галлюцинации как истинные, попытки переубеждения бессмысленны. Бред – это расстройство мышления, при котором больные страдают манией преследования, считают, что имеют особое происхождение и способности, бессмертность, безнаказанность и т.д.
Основные клинические симптомы болезни Гентингтона включают слабоумие, встречающееся у всех пациентов. Человек перестает выполнять обычную работу, не может сосредоточиться, постоянно забывает.
Ограниченная подвижность и слабоумие – одни из самых печальных проявлений, потому что они лишают человека самостоятельности на всю жизнь.
Выделяются 3 основных типа болезни Гентингтона:
- Ювенильная форма. Появляется к 20 годам. При этом типе непроизвольные движения обычно отсутствуют, но возникает мышечная скованность. Наблюдается быстрое развитие деменции, расстройств личности, речевых нарушений.
- Классическая форма. Проявляется в возрасте 35-50 лет и встречается в 90% случаев. Характеризуется типичным курсом, описанным выше.
- Форма позднего старта. Появляется после 60 лет. Из всех трех типов этот является одним из самых легких – его течение медленное, значительное слабоумие отсутствует, человек сохраняет самостоятельность.
Диагностика
Фундаментальное обследование – генетическое, доказывающее умножение триплетов CAG. В соответствии с уровнем мутаций оно определяет приблизительный возраст, в котором может проявиться расстройство. Такое тестирование предполагает соблюдение очень строгих этических правил. Только пациенты, считающие это целесообразным, полностью согласные с тестом, могут сдавать анализы. Совершенно недопустимо принуждать человека к тестированию, переубеждать его. Из-за невозможности лечения необходимо связать эти тесты с поддержкой психолога и, при необходимости, социальных работников.

Из вспомогательных исследований (а также в рамках дифференциальной диагностики) можно выполнить КТ или МРТ. Цель этих обследований – определение нарушения базальных ганглиев, особенно хвостатого ядра.
У пациентов с подозрением на болезнь Гентингтона проводится специальный тест. Он подтверждает или исключает подозрение со 100% достоверностью. Всегда необходима подпись пациента, фиксирующая ознакомление с правилами диагностики.
Предиктивный (предсимптомный и пренатальный) тест проводится у бессимптомных людей с заболеванием в семейной истории, желающих узнать, поражены ли они болезнью. Но генетический тест только подтверждает наличие мутации и предпосылок к болезни, а не сам диагноз.
При беременности у женщин, у которых развилась хорея Гентингтона, диагностика может заключаться в проведении теста, направленного на определение генетического статуса их будущего ребенка. Этот пренатальный тест выполняется путем отбора амниотической жидкости (амниоцентез).
Лица, желающие пройти предсимптомное тестирование, должны тщательно рассмотреть последствия. Они имеют право не знать свой генетический статус.
В связи с ответственностью, предсимптомное тестирование не проводится для лиц младше 18 лет.
Лечение
Болезнь Гентингтона в настоящее время неизлечима. Врачи пытаются противодействовать многим ее проявлениям. Заболевание требует сотрудничества специалистов из области неврологии, психологии, психиатрии, физиотерапии, трудотерапии, логопедии и др.
В очень серьезных случаях непроизвольных движений используются антипсихотические, успокоительные средства или нейролептики. Они также применяются в случаях бреда, галлюцинаций, агрессии, беспокойства. Однако такие лекарства имеют много побочных эффектов, поэтому находят применение только в действительно тяжелых случаях, после тщательной оценки возможных рисков.

В процессе лечения хореи Гентингтона важно бороться с потерей веса, принимая высококалорийную пищу (более 5000 ккал/день). Желательно обсудить питание с диетологом. Рекомендуется употреблять жирное мясо, цельные молочные продукты, жирную рыбу, сладкие соки, картофель, шоколад.
Против депрессии эффективны антидепрессанты. При нарушениях сна, вызванных беспокойством, по рекомендации врача принимаются снотворные препараты. Возможно также применение народных методов, например, успокаивающих травяных чаев.
К сожалению, самое серьезное проявление заболевания – слабоумие – совершенно неизлечимо. Его даже нельзя замедлить.
Осложнения
Одно из осложнений – инфекции. Наиболее распространенная инфекция – пневмония, способная быть смертельной для пациента из-за ослабленного иммунитета.
Сосудистые нарушения, закупорка артерий могут привести к сердечной недостаточности. Серьезное осложнение – расстройство глотания и приема пищи. Пациент сильно худеет, истощается.

Все остальные осложнения связаны с симптомами, относящимися к самой болезни. Особенно разрушительным является изменение в поведении. Часто человек становится эгоцентричным, агрессивным, навязчивым. Осложнение этого поведения – более высокая склонность к зависимости. Опасность представляют тенденции к самоубийству, которые при болезни Гентингтона намного выше, чем в среднем по населению.
Важно знать
Болезнь Гентингтона связана не только с самим пациентом, но и с его семьей, окружением. Самые большие опасения относятся к здоровью потомства – генетический риск очень высок. Также нелегко объяснить детям, что на самом деле происходит с их родителями. Почему мама ведет себя так странно, что делает со своими руками и т.д. Часто тема заболевания становится запретной в широкой семье или в обществе, что усугубляет психическое состояние пациента.
Жизнь партнера больного человека выворачивается наизнанку. Он постепенно теряет свое свободное время, которое должен посвятить, заботе о нем. У большинства людей, живущих в семье с больным, развивается депрессия, негативное отношение к миру, вспышки гнева, постоянная грусть и т.д. Нередки случаи пристрастия к алкоголю.
К сожалению, в нашей стране нет специализированного учреждения долгосрочного или краткосрочного лечения этого заболевания. В то же время социальная поддержка играет жизненно важную роль в оказании помощи пациентам и их семьям.
Читайте также:
