
Х сцепленная рецессивная спиноцеребеллярная атаксия
Авторы: Тецуо Ашизава и Гуанбинь Ся
Пер. с англ. Н.Д. Фирсовой (2018)
Вступление
Атаксия, определяемая как нарушение координации произвольных мышечных движений, является физическим признаком, а не самостоятельным заболеванием, поэтому всегда необходимо исследовать ее этиологию. Атаксия может быть главной жалобой пациента или одним из симптомов.
Атаксия обычно вызывается дисфункцией мозжечка или поражением вестибулярного или проприоцептивного афферентного входа в мозжечок. Атаксия может иметь скрытое начало с хроническим и медленно прогрессирующим клиническим течением (например, спиноцеребеллярные атаксии [SCA] генетического происхождения) или острое начало, особенно те атаксии, возникающие в результате инфаркта мозжечка, кровоизлияния или инфекции, которые могут быстро прогрессировать с катастрофическими последствиями. Атаксия также может иметь подострое начало из-за инфекционных или иммунологических расстройств, которые могут иметь ограниченное окно терапевтических возможностей.
Оперативная стратегия устранения излечимых причин атаксии может спасти жизнь пациенту и привести к хорошим долгосрочным результатам. Атаксия также может быть доброкачественной при симптоматических расстройствах (например, вестибулярный неврит). С развитием нейрогенетики диагностируется больше случаев мозжечковой атаксии, вызванной наследственными причинами, но многие спорадические атаксии, в том числе с хроническим и прогрессирующим течением, все еще остаются недиагностированными.
Симптомы и признаки атаксии
Симптомы и признаки связаны с расположением поражений в мозжечке.
Латерализованные поражения мозжечка вызывают ипсилатеральные симптомы, тогда как диффузные поражения мозжечка вызывают более общие симметричные симптомы. Поражения в полушарии мозжечка вызывают атаксию конечностей (аппендикулярную). Поражения червя вызывают атаксию туловища и походки, с относительным щажением конечностей. Вестибулоцеребеллярные поражения вызывают нарушение равновесия, головокружение и атаксию походки (т. н. мозжечковая походка).
Острая патология мозжечка может первоначально вызвать серьезные отклонения; со временем он может значительно восстановиться, и заболевание стать бессимптомным, даже если визуализация показывает постоянные драматические структурные изменения в мозжечке.
Хроническая прогрессирующая атаксия может быть связана не только с нейродегенеративными или наследственными заболеваниями мозжечка, но также с новообразованиями и хроническими инфекциями.
Термины, описывающие атаксию
Следующие клинические термины часто используются при описании атаксии.
Устойчивость. Здоровый человек может стоять естественным образом, с ногами, расставленными на расстояние менее 12 см друг от друга, и может стоять устойчиво, когда его ноги вместе, более 30 секунд. Нарушение позы при отсутствии двигательной слабости или грубых непроизвольных движений наводит на мысль о мозжечковой или сенсорной атаксии.
Атаксичная походка. Атаксия походки возникает в результате нарушения координации нижних конечностей из-за патологии мозжечка или потери проприоцептивного взаимодействия. Пациенты часто чувствуют себя неуверенно и вынуждены держаться за стену или мебель и ходить, расставив ноги. Усиливающееся нарушение походки при удалении зрительных сигналов (ходьба с закрытыми глазами или в темноте) указывает на сенсорный или вестибулярный компонент атаксии. Атаксия, вызванная мозжечковыми причинами, остается неизменной независимо от визуальных признаков.
Сенсорная атаксия. Сенсорная атаксия в основном проявляется нарушением походки, как описано выше. Кроме того, субъекты с сенсорной атаксией будут иметь положительный симптом Ромберга. Они могут ходить высокой походкой (из-за моторной слабости) или шаркая ногами (чтобы помочь себе обратной сенсорной связью, вызванной звуком). Псевдоатетоз (случайные движения пальцев, наблюдаемые на вытянутых руках с закрытыми глазами) также может возникать при сенсорной нейронопатии, поражающей верхние конечности.
Туловищная атаксия. Атаксия туловища может возникнуть в результате поражения средней линии мозжечка. Пациенты могут проявлять нестабильность в области туловища в виде колебаний тела сидя (хуже с вытянутыми вперед руками) или стоя (титубация).
Дисдиадохокинез / дисритмокинез. Дисдиадохокинез, или дисритмокинез проверяется путем быстрого чередования движений рук или постукивания указательным пальцем по складке большого пальца. Нарушения могут проявляться неравномерностью ритма и амплитуды.
Интенционный тремор. Интенционный тремор возникает в результате нестабильности проксимального отдела конечности и проявляется в увеличении амплитуды колебаний в конце произвольного движения. Проверяется прикосновением пальца к носу и пятки к голени. Это отличается от эссенциального тремора, который в основном происходит в дистальной части конечности.
Дисметрия. Дисметрия – это когда пациент пропускает целевой объект из-за превышения (гиперметрия) или из-за недостаточности (гипометрия) преодоления необходимого расстояния. Дисметрия часто проверяется с помощью теста погони за пальцем и может быть определена количественно по пропущенному расстоянию (в см). Дисметрия также бывает, когда глаза выключают предметы (глазная дисметрия); либо глазам нужно второе движение, чтобы поймать объект (гипометрические саккады), либо нужно скорректировать превышение, чтобы сфокусироваться на объекте (гиперметрические саккады). Тест на постукивание по голени (точное постукивание по середине голени или колена пяткой противоположной ноги) также обнаруживает наличие дисметрии.
Дизартрия. Дисартрия часто описывается пациентом или родственниками как невнятная речь. Речь пациента нерегулярна и медленна с ненужными колебаниями. Слова часто разбиты на отдельные слоги, а некоторые слоги с взрывным согласным необычно ударены (скандирующая речь).
Нистагм. Нистагм часто встречается при мозжечковых заболеваниях. Латеральный нистагм, вызванный боковым взглядом, виден медленным дрейфом к средней линии, за которым следует быстрая фаза саккад в эксцентрическом положении. Нистагм при взгляде вверх и нистагм при взгляде вниз определяются быстрой фазой в направлении вверх или вниз. Нистагм при взгляде вверх наблюдается при поражении передней части червя. Нистагм при взгляде вниз, как правило, проявляется при повреждении большого отверстия, например, при пороке развития Арнольда-Киари.
Саккады. Скорость саккад обычно нормальна при мозжечковом заболевании, но часто наблюдается чрезмерно далекий или слишком близкий взгляд (глазная дисметрия), и часто за ним следует корректирующая саккада в соответствующем направлении. Однако в SCA типа 2 (SCA2) и в продвинутых стадиях других SCA саккады замедляются.
Квадратноволновые толчки / окулярный флаттер / опсоклонус. Квадратноволновые толчки, окулярный флаттер и опсоклонус – это термины, используемые для описания других глазных нарушений при поражении мозжечка. Квадратноволновые толчки появляются в виде двух саккад в противоположных направлениях, разделенных коротким периодом отсутствия движения. У здорового человека может быть квадратноволновое движение от 0,1 до 0,3 градуса, но менее 10 в минуту. Квадратноволновые толчки с большой амплитудой более характерны для мозжечковой атаксии. Окулярный флаттер отличается от квадратноволновых толчков тем, что повторяющиеся саккады не разделены короткими периодами отсутствия движения. Опсоклонус – это сплошные сопряженные саккады во всех направлениях хаотичного характера. Как и окулярный флаттер, так и опсоклонус обычно указывают на поражение мозжечка при паранеопластическом (нейробластома) или постинфекционном синдроме (как можно видеть при атаксии опсоклонус-миоклонус).
Нейроанатомия атаксии
Мозжечок, его афферентные и эфферентные связи, вестибулярная система и проприоцептивный сенсорный путь – все они вовлечены в атаксию. Мозжечок состоит из срединной части и полушарий. Поражения в каждой из этих областей могут привести к различной форме атаксии. Например, повреждение срединных структур мозжечка обычно сопровождается атаксией походки и туловища, тогда как унилатеральное повреждение полушария мозжечка обычно вызывает ипсилатеральную мозжечковую атаксию. Понимание этой нейроанатомии и взаимосвязи с координацией может помочь с локализацией повреждения.
Классификация и этиология атаксии
Существуют различные способы классификации атаксии: по возрасту начала, темпу начала и клиническому течению; по анатомическому вовлечению; очаговое поражение или генерализованное; приобретенное или унаследованное. Случаи, когда все конкретные диагнозы были исключены, классифицируются как спорадическая атаксия у взрослых с неизвестной этиологией, которая все еще остается диагностической проблемой.
Наследственная атаксия
Наследственные атаксии относятся к редким заболеваниям, но по мере развития диагностических технологий они все чаще диагностируются в более раннем возрасте. Наследственные атаксии классифицируются как аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные или митохондриальные. Характерные особенности могут помочь в распознавании определенного диагноза.
Наследственные атаксии, особенно аутосомно-доминантные мозжечковые атаксии, следует учитывать, когда болезнь передается вертикально от одного поколения к другому в семье. Документация передачи от отца к сыну устанавливает аутосомно-доминантное наследование.
Спиноцеребеллярная атаксия
Наличие семейного анамнеза, согласующегося с аутосомно-доминантным наследованием, требует тестирования ДНК на наличие SCA, которая в генетической терминологии относится к группе аутосомно-доминантных нарушений с известным хромосомным локусом. Первичные митохондриальные и Х-связанные мутации также могут рассматриваться в зависимости от клинических проявлений и семейного анамнеза. При отсутствии семейного анамнеза и исключении вторичных атаксий спорадические случаи могут все еще требовать тестирования ДНК, поскольку отрицательный семейный анамнез не исключает наследственных расстройств, и до 5% пациентов с явно спорадической дегенеративной атаксией могут иметь положительный анализ ДНК. Из-за стоимости анализа ДНК, должно быть принято во внимание медицинское страхование. Хотя SCA неиз лечима, положительный результат анализа ДНК дает важную практическую информацию пациентам и их семьям. Во-первых, это позволяет пациентам планировать свое будущее, например, принимать решения об образовании или карьере, особенно в области прогнозного тестирования. Во-вторых, он обеспечивает основу для генетического консультирования пациентов и их семей. Наконец, это позволяет пациентам участвовать в группе поддержки и исследовательской деятельности, специфичной для генетически определенного заболевания. Например, генотип, определенный с помощью анализа ДНК, будет включен в критерии большинства, если не всех, будущих клинических испытаний болезнь-модифицирующих методов лечения, и как только терапия будет разработана и утверждена, пациентам необходимо будет знать генотип своего заболевания, чтобы определить подходит ли препарат для них.
Спорадическая и идиопатическая атаксия у взрослых
Для пожилых пациентов со спорадической дегенеративной атаксией неврологам следует рассмотреть диагноз мультисистемной атрофии мозжечкового типа (MSA-C). Документирование вовлечения других участков нервной системы, особенно базальных ганглиев и вегетативной дисфункции, имеет решающее значение для диагностики MSA-C у пациентов со спорадической атаксией. МРТ головного мозга может дать ключ к диагнозу MSA с атрофией ствола мозга. Оставшиеся дегенеративные атаксии с поздним началом часто называют спорадической атаксией у взрослых в категории, известной как идиопатическая мозжечковая атаксия с поздним началом. Хотя эти два термина используются взаимозаменяемо, некоторые эксперты включают MSA-C в категорию идиопатической мозжечковой атаксии с поздним началом.
Атаксия Фридрейх а
Атаксия Фридрейха является одним из наиболее распространенных генетических аутосомно-рецессивных атакси ческих синдромов. Начало обычно в детском и молодом возрасте с прогрессирующей атаксией, ведущей к потере передвижения после 10-15 лет. Другие клинические признаки включают потерю чувствительности из-за дорсального корешкового ганглия и дегенерации дорсального столба с арефлексией и деформациями стоп, сколиоз ом, гипертрофическ ой кардиомиопати ей и непереносимость ю глюкозы. При генетической диагностике фенотип расширился и теперь включает более позднюю картину (старше 25 лет и до 60 лет) и более медленное прогрессирование, которое называется атаксией Фридрейха с поздним началом. У таких людей сохраняются рефлексы, часто называемые атаксией Фридрейха с сохраненными рефлексами, которые могут обнаруживать спастичность без сердечных или скелетных признаков. Заболевание возникает из-за аномального расширения повторов GAA в гене фратаксина (FXN), хотя в редких случаях на одной из хромосом вместо расширения GAA могут присутствовать точечные мутации. Возраст начала заболевания обратно коррелирует с числом повторов GAA на меньшей аллели. Потеря функции фратаксина в митохондриях приводит к дефициту железо-серных кластеров, нарушению окисления и накоплению железа. Было показано, что никотинамид повышает уровень фратаксина, но клинической пользы пока не было продемонстрировано, и исследования с хелаторным деферипроном и антиоксидантом идебеноном неясны в отношении долгосрочной пользы. Были завершены клинические испытания с карбамилированным эритропоэтином, интерфероном гамма-1b, пиоглитазоном, эпоэтином альфа и некоторыми другими веществами. Эти исследования не показали убедительных доказательств эффективности.
OMIM 164400
Наша команда профессионалов ответит на ваши вопросы
Аутосомно-доминантые церебеллярные атаксии. В настоящее время известно более двадцати форм этого заболевания. Клиническая картина в основном определяется поражением мозжечка, иногда страдают также базальные ядра, ствол мозга, спинной мозг, зрительные нервы, сетчатка и спинномозговые нервы. Заболевание может проявляться только мозжечковыми нарушениями или их сочетанием с симптомами поражения перечисленных структур. Изредка развивается деменция. Первыми симптомами заболевания бывает незаметно появляющаяся неловкость, неустойчивость при быстрой ходьбе и беге. Через несколько лет у больного постепенно развивается развернутый атаксический синдром, отмечается неловкость и нарушение координации в руках, интенционный тремор конечностей, адиадохокинез (неритмичность, замедленность движений), скандировання речь, характерным образом нарушается почерк (макрография, неровность строк). Характерны симптомы вовлечения пирамидного и экстрапирамидного тракта, офтальмоплегия (паралич мышц глаза), атрофия зрительных нервов, пигментная дегенерация сетчатки, нистагм (непроизвольные ритмические судорожные движения глазного яблока), амиотрофии.
Молекулярно-генетической причиной заболеваний является увеличение числа тринуклеотидных САG повторов в кодирующем гене. Число повторов обратно пропорционально возрасту манифестации, и прямо пропорционально скорости развития и тяжести заболевания. Характерен феномен антиципации – утяжеление клинических проявлений заболевания из поколения в поколение в пределах одной родословной (более раннее начало и быстрое прогрессирование заболевания, проявление более тяжелых симптомов у потомков). Феномен антиципации обусловлен нестабильностью повтора и нарастанием его длины при передаче мутантного гена от родителя потомкам. Эффект “отцовской передачи” (манифестация более ранних и более тяжелых случаев болезни у потомков больного отца) – обусловлен преимущественным удлинением мутантного повтора в мужском гаметогенезе, тогда как при передаче гена от матери область повтора обычно остается стабильной.
В Центре Молекулярной Генетики проводится прямая молекулярно-генетическая диагностика наиболее частых форм спиноцеребеллярной атаксии: SCA 1, 2, 3, 6, 7, 8, 12 и 17 типов, которая основана на оценке числа CAG-повторов, локализованных в генах ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, ATXN8, PPP2R2B и TBP .
Спиноцеребеллярная атаксия 1 (SCA 1, OMIM 164400).
Заболевание обычно начинается в возрасте от 30 до 40 лет (возможный разброс – от 4 до 74 лет). Основные клинические симптомы – атаксия, офтальмоплегия, пирамидные и экстрапирамидные расстройства.
Молекулярно-генетической причиной SCA1 является увеличение числа тринуклеотидных САG повторов в гене ATXN1, располагающемся на 6-й хромосоме (сегмент 6р23). Длина гена составляет 450000 нуклеотидов. Ген содержит девять экзонов. Транскрипт состоит из 10660 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их меньше 36; при болезни больше 40). Нормальные аллели гена обычно содержат в составе тринуклеотидного участка вставки от одного до трех CAT-триплетов, отсутствующие в мутантном гене. Их наличие рассматривается как важный фактор стабилизации нормальных аллелей при мейозе.
Спиноцеребеллярная атаксия 2 (SCA 2, OMIM 183090).
Заболевание обычно начинается в возрасте от 20 до 40 лет (возможный разброс – от 6 до 67 лет). Основные клинические симптомы – атаксия, замедление саккадических движений глазных яблок (саккады - скачкообразные быстрые содружественные фиксирующие движения глаз, возникают, когда взгляд переводится с одного неподвижного предмета на другой), пирамидные и экстрапирамидные расстройства.
Молекулярно-генетической причиной SCA2 является увеличение числа тринуклеотидных САG повторов в гене ATXN2, располагающемся на 12-й хромосоме (сегмент 12q24). Ген содержит 25 экзонов. Длина гена составляет 130000 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их 14-31; при болезни 35-64). Нормальные аллели гена обычно содержат в составе тринуклеотидного участка отдельные вставки CAА-триплетов, стабилизирующих повтор.
Спиноцеребеллярная атаксия 3 (SCA3, болезнь Мачадо-Джозефа, OMIM 109150)
Заболевание обычно начинается после 25-30 лет (возможный разброс – от 5 до 70 лет). Основные клинические симптомы - атаксия (в первую очередь наблюдается абазия (нарушение походки)) офтальмоплегия, парез взора вверх, фасцикуляции мышц лица и фасцикуляции мышц языка, феномен “выпученных глаз” (широко раскрытые глазные щели с фиксированными глазными яблоками), пирамидные и экстрапирамидные симптомы (паркинсонизм). Молекулярно-генетической причиной SCA3 является увеличение числа тринуклеотидных САG повторов в гене ATXN3, располагающемся на 14 хромосоме (сегмент 14q24.3-q31). Ген содержит 11 экзонов. Длина гена составляет 48200 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их 12-47; при болезни 53-86)
Спиноцеребеллярная атаксия 6 (SCA6, OMIM 183086)
Спиноцеребеллярная атаксия 6 типа – аутосомно-доминантное заболевание, частота которого 3:100000. Первый признак - нарушение походки, которое в конечном итоге приковывает пациента к инвалидному креслу. Обычно SCA6 начинается в более позднем возрасте по сравнению с 1-3 типами. Основные клинические симптомы - мягкая прогрессирующая атаксия, нистагм при фиксировании взгляда, дизартрия, дисфагия. При МРТ обнаруживается изолированная атрофия мозжечка. Молекулярно-генетическая причина SCA6 – небольшаяэкспансия тринуклеотидных CAG-повторов, находящихся в 3’ кодирующей области гена CACNA1A, расположенном на хромосоме 19(сегмент 19p13). В норме количество повторов от 5 до 20, при болезни обнаруживается от 21 до 25. Ген CACNA1A кодирует CACNA1А порообразующую субъединицу кальциевого канала, эксперссирующуюся преимущественно в мозжечке (в гранулярных клетках и клетках Пуркинье). Мутации в этом гене приводят к развитию двух других заболеваний – эпизодической атаксии второго типа и семейной гемиплегической мигрени.
Спиноцеребеллярная атаксия 7 (SCA7, OMIM 164500)
Спиноцеребеллярная атаксия 7 типа (оливопонтоцеребеллярная атрофия 3 типа) – прогрессирующее аутосомно-доминантное нейродегенеративное заболевание, клинически характеризующееся церебеллярной атаксией, ассоциированной с дистрофией желтого пятна. Средний возраст манифестации заболевания – 32 года. Степень тяжести, скорость прогрессии и возраст начала заболевания варьируют как между семьями так и внутри семей. Основные клинические симптомы – офтальмоплегия, пирамидные и экстрапирамидные знаки, дизартрия, дисфагия, хорея, гиперрефлексия, спастика, потеря глубокой чувствительности, пигментная дегенерация сетчатки, прогрессирующая потеря зрения, медленные саккады, атрофия зрительного нерва.
Молекулярно-генетическая причина SCA7 –экспансия тринуклеотидных CAG-повторов гена ATXN7 (3p21.1-p12), находящихся в полиглутаминовом тракте белка ataxin-7.В норме количество повторов варьирует от 4 до 35, при болезни обнаруживается от 36 до 306 повторов.
Спиноцеребеллярная атаксия 8 (SCA8, OMIM 608768)
Спиноцеребеллярная атаксия 8 типа – медленно прогрессирующее аутосомно-доминантное заболевание, возраст начала которого варьирует от 18 до 65 лет. Основные клинические симптомы – прогрессирующая церебеллярная атаксия, нарушение координации походки, движения конечностей, речи, брадикинезия (замедленные движения). У больных часто наблюдается дизартрия, тремор, дисфагия, нистагм, замедление саккадических движений глазных яблок, дизметрические саккады, потеря чувствительности. При МРТ обнаруживается атрофия полушарий и червя мозжечка. Молекулярно-генетической причиной SCA8 является увеличение числа тринуклеотидных СAG повторов в гене ATXN8. В норме количество повторов варьирует от 15 до 50, при болезни обнаруживается от 71 до 1300.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Спиноцеребеллярная атаксия 12 (SCA12, OMIM 604326)
Спиноцеребеллярная атаксия 12 типа – аутосомно-доминантное заболевание. Начинается в возрасте от 8 до 55 лет. Основные клинические симптомы - тремор верхних конечностей, тремор головы, атаксия, дисметрия, дисдиадохокинез, гиперрефлексия, скудость движения, аномальные движения глаз, деменция. При МРТ и КТ обнаруживается атрофия коры головного мозга и мозжечка.
Молекулярно-генетическая причина SCA12 – увеличение числа тринуклеотидных CAG повторов в гене PPP2R2B, распологающемся на 5-ой хромосоме (сегмент q31-q33). В норме количество повторов от 7 до 32, при болезни обнаруживаются от 51 до 78. Клиническое значение экспансии повторов в диапазоне от 33 до 50 до сих пор не установлено. Ген PPP2R2B кодирует регуляторную субъединицу В белка фосфатазы 2 участвующего в таких регуляторных процессах, как рост и деление клеток, сокращение мышц и транскрипция генов.
Спиноцеребеллярная атаксия 17 (SCA 17, OMIM: 607136)
Спиноцеребеллярная атаксия 17 типа – аутосомно-доминантное неврологическое расстройство, характеризующееся атаксией, пирамидными и экстрапирамидными расстройствами (паркинсонизм), когнитивными нарушениями, психозом и судорогами. Клинические проявления схожи с Хореей Гентингтона. Заболевание обычно начинается в возрасте от 23 до 29 лет (возможный разброс - от 3 до 53 лет).
Молекулярно-генетической причиной SCA17 является увеличение числа тринуклеотидных CAG (или CAA) повторов в гене TBP, расположенном на длинном плече 6 хромосомы (сегмент q27). Ген TBP кодирует фактор транскрипции-белок, связывающий последовательность ТАТА-бокса (ТВР). В норме количество повторов варьирует от 25 до 44, при болезни обнаруживается от 47 до 63 повторов. Описана связь 45 и 46 повторов с неполной пенетрантностью заболевания.
а) Атаксия Фридрейха. Атаксия Фридрейха — наиболее четко описанная и часто встречающаяся спиноцеребеллярная дегенерация. Частота встречаемости гена составляет 1:110 человек в Англии (Harding 1981a), и примерно один из 10000 человек в Швеции имеет клинические проявления.
Ген атаксии Фридрейха включает повторы ГА А последовательности в интроне 1, который распространен у пациентов (120-1700 повторов). Продуктом нормального гена является белок фратаксин, функция которого не полностью ясна. 94% пациентов с типичной атаксией Фридрейха являются гомозиготами по ГАА экспансии, тем не менее продолжительность повтора на каждой хромосоме из пары неодинакова (Durr et al., 1996a).
В редких случаях отмечается только одна мутация, но в такой ситуации выявляется точечная мутация в гомозиготном локусе (Campuzano et al., 1996). Выраженная длина повтора коррелирует с началом в раннем возрасте, более стремительным течением и наличием кардиомиопатии (Durr et al., 1996a).
Второй ген на хромосоме 9p23-p11 является причиной редких случаев (Фридрейха 2), клинически нечетко отличаемых от 1 типа (Christodoulou et al., 2001).
Основным патологическим проявлением является дистальная аксональная нейропатия, которая поражает нейроны длинных восходящих и нисходящих трактов спинного мозга и крупные сенсорные волокна периферических нервов и ганглии задних корешков (Said et al., 1986). Также зарегистрирована утрата нервных волокон в зрительных путях, а мозжечок остается непораженным.
Сердце увеличено, и более чем в половине случаев отмечается гипертрофическая кардиомиопатия с некрозом волокон и фиброзом, преимущественно затрагивающим левый желудочек.
Критерии диагностики атаксии Фридрейха (Harding, 1981a) включают начало до 25 лет (обычно до 16 лет), аутосомно-рецессивное наследование и сочетанное поражение крупных сенсорных волокон периферических нервов, мозжечкового тракта, пирамидного тракта и задних столбов.
Тем не менее, степень фенотипической вариабельности велика, в некоторых случаях отмечается позднее начало и/или меньшая выраженность симптомов и вариабельное течение, и некоторые пациенты прикованы к инвалидной коляске в раннем подростковом возрасте, в то время как другие способны самостоятельно передвигаться почти до 40 лет (Montermini et al., 1997).
По неофициальным данным, к доминантным случаям относится большая часть наследственной моторной и сенсорной нейропатии со скелетными деформациями и утратой чувствительности, но некоторые случаи не поддаются классификации.

Клинические проявления атаксии Фридрейха были описаны у 115 пациентов из 90 семей (Harding, 1981b). Основные проявления представлены в таблице ниже.
Заболевание чаще всего начинается в возрасте 5-16 лет, в редких случаях — в возрасте 2-5 лет. Прогрессирующая атаксия нижних конечностей с нарушением походки является основным проявлением, в то время как поражение верхних конечностей, приводящее к неуклюжести, в ранние сроки отмечается только в 25% случаев.
Сколиоз, тремор и изменения со стороны сердца редко являются первыми проявлениями, но формируются со временем, особенно при раннем начале заболевания. Конская стопа также является ранним симптомом. При осмотре в 70-95% случаев выявляется отсутствие глубоких рефлексов.
Дизартрия, пирамидные знаки со стороны ног и утрата глубокой и вибрационной чувствительности могут появляться позже, но относятся к постоянным симптомам. Нистагм встречается нечасто (20% случаев), медленные изломанные следящие движения глаз выявляются в 12% случаев (Harding, 1981b). Нестабильность фиксации является типичным проявлением (Alper и Narayanan, 2003).
Нередко встречается атрофия зрительного нерва, а глухота отмечается только у 10% пациентов. Дистальная атрофия отмечается практически в половине случаев. Интеллект не страдает.
Поражение сердца по результатам ЭКГ обнаруживается в трети случаев и даже чаще, если ЭКГ проводится систематически. Изменения зубца Т и аномалии сегмента ST являются ранними признаками сердечной недостаточности и единственным ее проявлением.
В конечном счете формируется прогрессирующая сердечная недостаточность или аритмия с фибрилляцией предсердий, половина пациентов умирает от сердечной недостаточности (Leone et al., 1988).
Течение заболевания медленное, но прогрессирующее. В среднем пациенты утрачивали способность ходить к 25 годам, со средней продолжительностью заболевания 15,5 лет.
Сахарный диабет является дальнейшим осложнением и развивается у 10% пациентов. Он имеет тенденцию сочетаться с атрофией зрительного нерва, в некоторых случаях диабетическая кома является причиной смерти.
Атипичные формы включают легкие случаи, которые, возможно, связаны с одним и тем же локусом 9-й хромосомы. К данной группе относятся случаи сохранения сухожильных рефлексов (Palau et al., 1995) и поздние формы с началом в раннем взрослом возрасте (De Michele et al., 1994).
Результаты одной из недавних работ, в которой использовалось возможное выявление мутантного гена, предполагают, что клиническая картина более вариабельна, чем считалось раньше (Palau et al., 1995; Pandolfo 2003). 25% пациентов в рамкам одного крупного исследования имели одно или более атипичное проявление (начало после 25 лет, сохранение или даже оживление сухожильных рефлексов или отсутствие симптома Бабинского).
Возраст начала превышал 20 лет у 19 из 114 пациентов (De Michele et al., 1994). Сохраненные рефлексы среди пациентов, которые в остальном соответствуют всем критериям, зарегистрированы у значимого числа пациентов (Palau et al., 1995). Большая часть случаев, ранее отнесенных к рано начинающейся атаксии с сохранением глубоких сухожильных рефлексов (состояния, отличного от атаксии Фридрейха) (Harding, 1981b; Klockgether et al., 1991), по результатам молекулярно-генетических исследований, вероятно, имеют отношение к болезни Фридрейха.
Эта группа, очевидно, была гетерогенной как при раннем ( 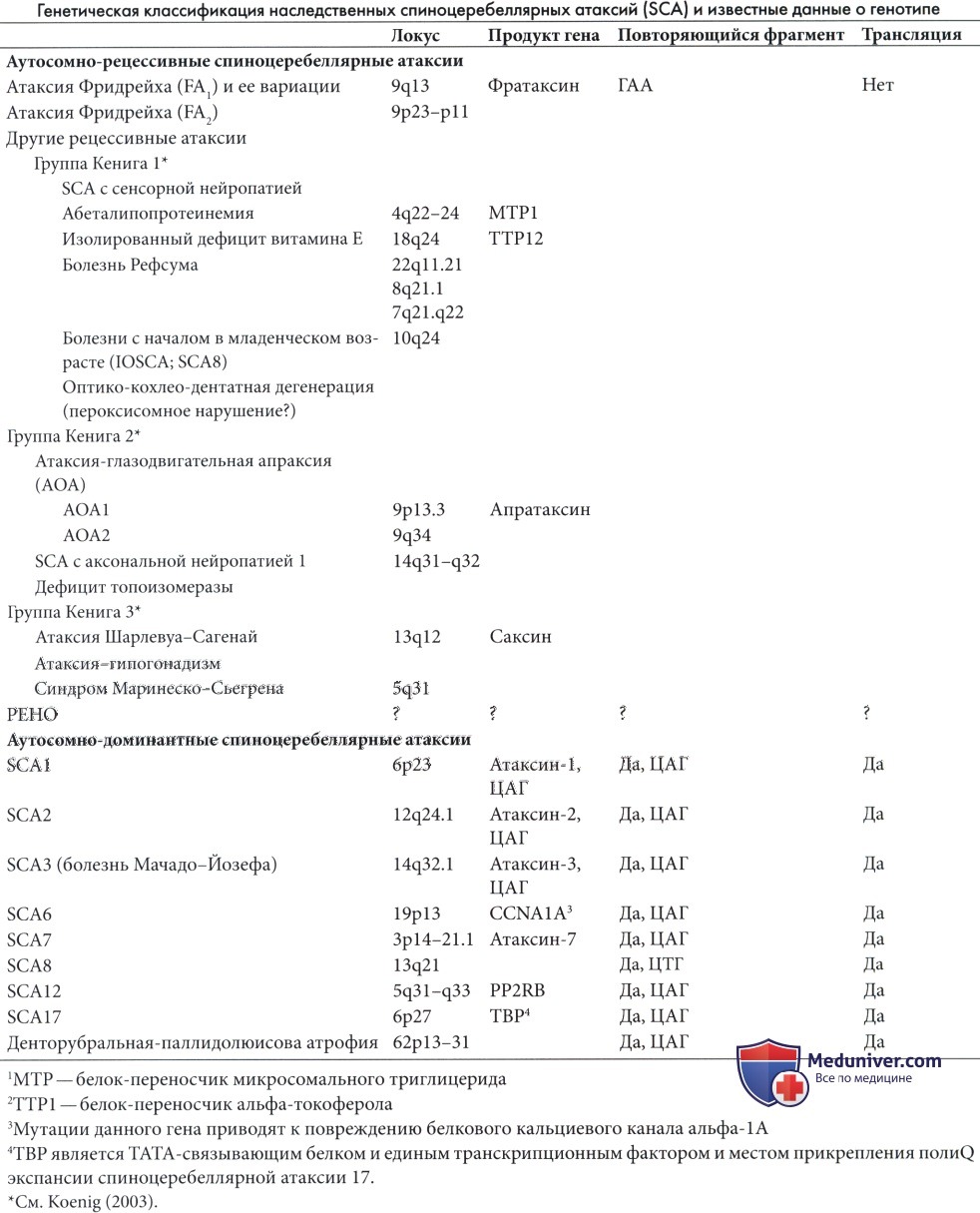
Вторая подгруппа Кенига включает атаксию со зрительной моторной апраксией (АОА), которая делится на два типа: AOA1 является одной из наиболее распространенных форм детского возраста и описана вместе с атаксией-телеангиэктазией, несмотря на то, что ее физиология кажется более сходной с спиноцеребеллярными атаксиями (SCA, см. далее). Одним из важных биологических признаков является гипоальбуминемия, которая практически постоянно обнаруживается и имеет диагностическую значимость. АОА 2 типа встречается реже и начинается позже (в позднем подростковом или раннем взрослом возрасте). Умеренно повышенный уровень альфа-фетопротеина отмечается в 75% случаев.
Клинические проявления AOA1 очень напоминают проявления атаксии-телеангиэктазии, но без признаков экстраневрологических поражений.
В отличии от атаксии-телеангиэктазии, AOA1 не связана с повышением уровня альфа-фетопротеина, хромосомными аномалиями, склонностью к раковым опухолям или повышенной радиочувствительностью культуры фибробластов (Le Ber et al., 2005). Редким, но интересным состоянием является спиноцеребеллярная атаксия с аксональной нейропатией 1 (SCA1), которая фактически является нарушением репарации ДНК, вызванной отсутствием фермента топоизомеразы-фосфодиэстеразы-1 (TDP1) (E1-Khamisy et al., 2005).
В третьей подгруппе Кенига четко описана атаксия Шарлевуа-Сагеней. Изначально синдром был описан в Квебеке, но с тех пор регистрировался и в других частях света (Gucuyener et al., 2001). Заболевание связано с мутацией гена сакцина на 13-й хромосоме (Engert et al., 2000). Фенотипические проявления включают заметную спастичность и постоянное наличие полос на глазном дне с преобладанием миелиновых волокон, радиально расходящихся от диска зрительного нерва.
Два редких аутосомно-рецессивных синдрома включают очень медленно прогрессирующую атаксию и могут рассматриваться вместе с SCA. Несмотря на то, что патология и механизмы заболеваний отличаются, они проявляются несколькими общими клиническими симптомами.
Синдром Маринеску-Шегрена включает атрофию мозжечка, преимущественно затрагивающую червь, раннее начало медленно прогрессирующей атаксии, катаракту, легкую задержку умственного развития, иногда гипогонадизм (Sewry et al., 1988) и позднее развитие специфической миопатии (Superneau et al., 1987). Заболевание развивается в результате мутации гена SLI1, кодирующего белок-шаперон, ключевой регулятор основных функций эндоплазматической сети.
РЕНО синдром (прогрессирующая энцефалопатия с периферическими отеками, гипсаритмией и атрофией зрительного нерва, также называемая церебелло-оптический синдром) является рецессивным заболеванием, зарегистрированным преимущественно в Финляндии (Salonen et al., 1991), хотя регистрировались случаи и в других странах. Основными проявлениями являются рано начинающиеся припадки, легкий дисморфизм, периферические отеки и регрессия, начинающаяся в возрасте 3-5 месяцев. Атрофия зрительного нерва развивается к концу первого года (Haltia и Somer, 1993).
- Вернуться в оглавление раздела "Неврология."
Редактор: Искандер Милевски. Дата публикации: 18.12.2018
Читайте также:
