
Наследственные моторно-сенсорные невропатии шарко-мари-тута
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив - Клинические протоколы МЗ РК - 2010 (Приказ №239)
Общая информация
Протокол "Наследственная моторная и сенсорная невропатия"
Коды по МКБ-10: G 60,0
- Болезнь Шарко-Мари Тута (наследственная мотосенсорная невропатия, тип I)
- Болезнь Дежерина - Сотта (наследственная мотосенсорная невропатия, тип III)
- Наследственная моторная и сенсорная невропатия, типы I-IV
- Гипертрофическая невропатия у детей
- Перонеальная мышечная атрофия (аксональный тип, гипертрофический тип)


Классификация
Клинико-генетическая классификация наследственных мотосенсорных невропатий
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип I:
- обусловленная дупликацией 17 р 11.2;
- обусловленная точковыми мутациями гена миелина РМР-22.
Тип I В: обусловленная точковыми мутациями гена миелина Ро.
Тип I С: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип I: с неидентифицированным генетическим дефектом.
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Наследственная мотосенсорная невропатия, тип III:
- обусловленная дупликацией 17 р 11.2;
- обусловленная точковыми мутациями гена миелина РМР-22;
- обусловленная точковыми мутациями гена миелина Ро.
Х- сцепленная наследственная мотосенсорная невропатия:
- обусловленная точковыми мутациями и делециями коннексина 32;
- с неидентифицированным генетическим дефектом.
Сложные формы наследственных мотосенсорных невропатий:
- с пирамидными симптомами;
- с атрофией зрительных нервов;
- с атрофией зрительных нервов и глухотой;
- с пигментной ретинопатией;
- с кератодермией ладоней и стоп.
Диагностика
Диагностические критерии
В анамнезе: при болезни Шарко-Мари - аутосомно-рецессивный тип наследования, дебют заболевания в первое десятилетие жизни.
При болезни Дежерина-Сотта (наследственной мотосенсорной невропатии, тип III): аутосомно-рецессивный тип наследования, дебют в первые два года жизни, задержка моторного развития, слабость и атрофии дистальных отделов конечностей, по мере прогрессирования - поражение проксимальных отделов, атаксия, быстропрогрессирующее течение.
Для наследственной мотосенсорной невропатии, тип I характерен аутосомно-доминантный тип наследования, дебют заболевания преимущественно в 1-2 десятилетие жизни, медленно-прогрессирующий характер течения.
Для наследственной мотосенсорной невропатии, тип II - характерен аутосомно-доминантный тип наследования, дебют во второй декаде жизни, отсутствие либо незначительно выраженные атрофии дистальных отделов конечностей, относительно доброкачественное течение.
Для наследственной мотосенсорной невропатии, тип II - характерен аутосомно-рецессивный тип наследования, дебют заболевания в раннем возрасте, выраженные атрофии и слабость дистальных отделов конечностей, деформации кистей и стоп, быстропрогрессирующий характер течения.
Лабораторные исследования: общий анализ крови и мочи без патологии.
Инструментальные исследования
ЭМГ различна: для наследственной мотосенсорной невропатии, тип I характерно снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Шарко-Мари снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Дежерина-Сотта значительное снижение (менее 12 м/с) скорости проведения импульса по периферическим нервам.
Для наследственной мотосенсорной невропатии, тип II (аутосомно-рецессивная форма) на ЭМГ характерно значительное снижение скорости проведения импульса по периферическим нервам (менее 38 м/с).
Для наследственной мотосенсорной невропатии, тип II (аутосомно-доминантная форма) умеренное снижение скорости проведения импульса по периферическим нервам.
Показания для консультации специалистов:
Минимум обследования при направлении в стационар:
1. Общий анализ крови и мочи.
4. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
Дополнительные диагностические мероприятия:
Дифференциальный диагноз
Дифференциально-диагностические признаки аутосомно-доминантных наследственных мотосенсорных невропатий
Аутосомно-доминантная наследственная мотосенсорная невропатия
Наша команда профессионалов ответит на ваши вопросы
Болезнь Шарко-Мари-Тута (ШМТ), или невральная амиотрофия Шарко-Мари, известная и как наследственная моторно-сенсорная невропатия (НМСН), - обширная группа генетически гетерогенных заболеваний периферических нервов, характеризующаяся симптомами прогрессирующей полинейропатии с преимущественным поражением мышц дистальных отделов конечностей. НМСН являются не только самым частым среди наследственных заболеваний периферической нервной системы, но и одним из самых частых наследственных заболеваний человека. Частота всех форм НМСН варьирует от 10 до 40:100000 в различных популяциях.
Клинико-генетическая гетерогенность невральной амиотрофии Шарко-Мари, явилась основанием для поиска локусов, сцепленных с данными заболеваниями. К настоящему времени картировано более 40 локусов, отвечающих за наследственные моторно-сенсорные нейропатии, идентифицировано более двадцати генов, мутации в которых приводят к развитию клинического фенотипа НМСН. Описаны все типы наследования НМСН: аутосомно-доминантный, аутосомно-рецессивный и Х-сцепленный. Наиболее часто встречается аутосомно-доминантное наследование.
Все моторно-сенсорные нейропатии в настоящее время по электронейромиографическим (ЭНМГ) и морфологическим признакам принято разделять на три основных типа: 1) демиелинизирующий (НМСНI), характеризующийся снижением скорости проведения импульса (СПИ) по срединному нерву, 2) аксональный вариант (НМСНII), характеризующийся нормальной или несколько сниженной СПИ по срединному нерву, 3) промежуточный вариант (intermedia) со СПИ по срединному нерву от 25 до 45 м/с. Величина СПИ, равная 38 м/с, определяемая по двигательной компоненте срединного нерва, считается условной границей между НМСНI (СПИ 38м/с). Таким образом, ЭНМГ исследование приобретает особый смысл для ДНК-диагностики, поскольку позволяет выделить наиболее оптимальный алгоритм генетического обследования для каждой семьи.
Возраст начала заболевания, его тяжесть и прогрессирование зависят от типа нейропатии, но могут сильно варьировать даже в пределах одной семьи. Наиболее часто встречается форма болезни НМСНIА - от 50% до 70 % всех случаев НМСН 1 типа в различных популяциях. В 10% случаев выявляются Х-сцепленные формы НМСН, среди которых преобладает форма с доминантным типом наследования – НМСНIX, составляющая 90% от всех Х-сцепленных полинейропатий. Среди НМСН II типа наиболее часто встречается доминантная форма – НМСНIIA – в 33% всех случаев (табл. 1).
Наследственные моторно-сенсорные нейропатии с аутосомно-рецессивным типом наследования сравнительно редки, но клинически не отличимы от НМСН с аутосомно-доминантным типом наследования. НМСН 4D(Lom), 4С, 4H и 4J являются одними из таких болезней. Примечательно, что в генах NDRG1 и SH3TC2 локализованы частые мутации, характерные для цыган.
Таблица 1. Гены, ответственные за развитие различных форм НМСН. (Синим цветом выделены гены, анализ которых проводится в ООО "Центр Молекулярной Генетики")

Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
МКБ-10

- Причины
- Патогенез
- Классификация
- Симптомы
- Осложнения
- Диагностика
- Лечение невральной амиотрофии Шарко-Мари-Тута
- Медикаментозная терапия
- Немедикаментозная терапия
- Хирургическое лечение
- Экспериментальное лечение
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание – приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Диагностика
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
- ЭНМГ. При электронейромиографии отмечаются признаки аксональной и демиелинизирующей нейропатии – замедление скорости проведения импульса по двигательным нервам, падение амплитуды М-ответов.
- Компьютерная паллестезиометрия. Данная диагностическая процедура позволяет объективно оценить снижение вибрационной чувствительности – наиболее ранний признаки болезни ШМТ.
- Гистология. При гистологическом исследовании биоптата большеберцового нерва обнаруживаются уменьшение количества миелиновых волокон, разрастание соединительнотканных волокон, атрофию миелина.
- ДНК-анализ. Подтверждающий метод исследования, верифицирующий диагноз. Выявляются дупликации гена белка периферического миелина (PMP22) на 17-й хромосоме.
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру – баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Комплексное проведение данных мероприятий позволяет увеличить мышечную силу, исправить нарушения равновесия и походки. Благодаря этому удается повысить бытовую, социальную адаптацию, работоспособность пациентов.
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства – метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 – ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута – тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Этиология и встречаемость болезни Шарко-Мари-Тута. Болезнь Шарко-Мари-Тута — генетически разнородная группа наследственных нейропатий, характеризующихся хронической моторной и сенсорной полинейропатией. Данное заболевание подразделяют согласно типам наследования, неврологическим изменениям и клиническим симптомам.
По определению, болезнь Шарко-Мари-Тута 1-го типа — аутосомно-доминантная демиелинизирующая нейропатия; ее распространенность приблизительно 15 на 100 000, она также генетически разнородна. Болезнь Шарко-Мари-Тута 1А типа, представляющая 70-80% всех случаев патологии, вызывается повышенной дозой белка PMP22 из-за дупликации гена PMP22 в хромосоме 17. Новые дупликации составляют 20-33% случаев болезни Шарко-Мари-Тута 1А типа; более 90% этих мутаций возникает в ходе мужского мейоза.
Белок PMP22 — внутримембранный гликопротеид. В периферической нервной системе PMP22 обнаруживают в только в компактном миелине. Функция PMP22 объяснена не полностью, но имеются подтверждения, что он играет ключевую роль в уплотнении миелина.
Доминантные мутации с утратой функции в гене PMP22 и увеличение дозы PMP22 вызывают демиелинизирующую периферическую полинейропатию. Увеличение дозы белка PMP22 возникает при тандемной дупликации участка р11.2 в хромосоме 17. Этот участок размером 1,5 мегабазы ограничен повторяющимися последовательностями ДНК, идентичными почти на 98%. Нарушение спаривания фланговых повторяющихся элементов в ходе мейоза может приводить к неравному кроссинговеру и образованию одной хроматиды с дупликацией региона в 1,5 мегабазы и другой с реципрокной делецией. [Реципрокная делеция вызывает наследственную нейропатию с параличами сдавливания].
Индивидуум, унаследовавший хроматиду с дупликацией, будет иметь три копии нормального гена PMP22 и, таким образом, избыточную экспрессию белка PMP22.
Избыточная экспрессия белка PMP22 или экспрессия его доминантных отрицательных форм приводит к невозможности формирования и поддержки компактного миелина. Образцы биопсии нервов от сильно пораженных детей демонстрируют рассеянную скудность миелина, в биоптатах нервов пациентов при меньшей степени поражения видны участки демиелинизации и гипертрофии миелиновой оболочки. Механизм образования данных изменений при избыточной экспрессии белка PMP22 остается неясным.
Слабость и атрофия мышц при болезни Шарко-Мари-Тута 1-го типа происходят в результате их денервации, вызванной дегенерацией аксонов. Длительные наблюдения за больными показывают возраст-зависимое уменьшение плотности нервных волокон, согласующееся с развитием симптомов болезни. Кроме того, исследования на мышиных моделях указывают, что миелин необходим для функционирования цитоскелета аксонов. Как демиелинизация изменяет цитоскелет аксонов и влияет на их дегенерацию, полностью не объяснено.
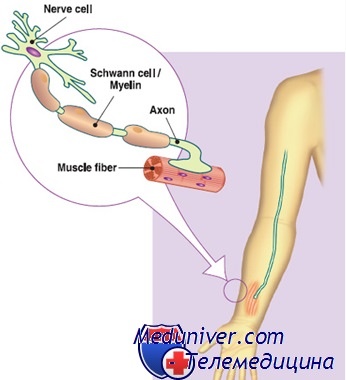
Болезнь Шарко-Мари-Тута 1А типа имеет почти полную пенетрантность, хотя тяжесть, возраст начала и течение болезни заметно изменяются внутри семей и между семьями. Большинство больных не обращаются за медицинской помощью, или не замечая симптомов, или поскольку эти симптомы легко переносятся. С другой стороны, многие имеют тяжелую болезнь, обнаруживаемую в раннем детстве.
Симптомы болезни обычно появляются в первые два десятилетия жизни; начало после 30 лет бывает редко. Обычно симптомы начинаются с незаметной медленно прогрессирующей слабости и атрофии дистальных мышц ног и легкого сенсорного ухудшения. Слабость в стопах приводит к аномалии походки, хлопающей стопе и, в конечном счете, изменению формы стопы (полая стопа молокообразные пальцы стоп) и утрате равновесия; но это редко приводит к утрате способности ходить.
Слабость важных мышц рук обычно появляется в конце болезни и, в серьезных случаях, вызывает аномалию кисти в виде сжатого кулака из-за дисбаланса между мышечной силой сгибателей и разгибателей кисти. Другие связанные симптомы — снижение или отсутствие рефлексов, атаксия и тремор верхних конечностей, сколиоз и пальпаторно утолщенные поверхностные нервы. Иногда также поражаются диафрагмальный и автономный нервы.
При электрофизиологических исследованиях патогномоничный признак болезни Шарко-Мари-Тута 1А типа — равномерное снижение скорости проведения во всех нервах и нервных сегментах в результате демиелинизации. Выраженное снижение скорости проведения обычно присутствует уже к 2-5 годам жизни, хотя клинически явные симптомы могут не обнаруживаться в течение многих лет.
Особенности фенотипических проявлений болезни Шарко-Мари-Тута:
• Возраст начала: от детства до зрелого возраста
• Прогрессирующая дистальная слабость
• Дистальная гипотрофия мышц
• Гипорефлексия
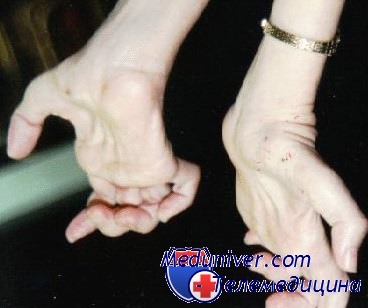
Хотя болезнь Шарко-Мари-Тута 1-го типа можно заподозрить на основе клинических, электрофизиологических и патогистологических признаков, окончательный диагноз часто зависит от обнаружения мутации. Воспалительные периферические нейропатии часто трудно отличить от болезни Шарко-Мари-Тута 1-го типа и наследственной нейропатии с параличами сдавливания, и ранее, до появления молекулярной диагностики, многих больных с унаследованной нейропатией лечили иммуносупрессорами, в итоге получая повышенную смертность без улучшения нейропатии.
Лечение симптоматическое, поскольку радикальной терапии к настоящему времени не разработано. Параллельно развитию болезни терапия обычно состоит из трех этапов: укрепляющие и растягивающие упражнения для поддержания ходьбы и других двигательных функций, использование ортопедической обуви и специальных шин и ортопедическая хирургия. При дальнейшем ухудшении могут понадобиться передвижные опоры, такие как костыли, ходунки или, в редких тяжелых случаях, инвалидное кресло. Всем больным необходимо рекомендовать избегать воздействия нейротоксичных медикаментов и химических веществ.
Поскольку дупликация и большинство точковых мутаций в гене PMP22 аутосомно-доминантные и полностью пенетрантные, каждый ребенок больного родителя имеет 50% шанс развития болезни Шарко-Мари-Тута 1А типа. Тем не менее переменная экспрессивность дупликации и мутаций в гене PMP22 делает невозможным прогноз тяжести болезни.
Пример болезни Шарко-Мари-Тута. В течение нескольких последних лет 18-летняя женщина обращала внимание на прогрессирующую слабость, снижение выносливости и способности бегать и ходить. Она также жаловалась на частые судороги ног, усиливающиеся при охлаждении, и появившиеся затруднения при перешагивании через предметы или подъеме по ступенькам. Она не помнила предшествующих болезней или жалоб, напоминающих воспалительный процесс, например мышечных болей, лихорадки, или ночных потов.
Скорость проведения по нервам оказалась аномальной; средний показатель составил 25 м/с (в норме >43 м/с). Результаты последующей биопсии нерва показали сегментарную демиелинизацию, гипертрофию миелиновой оболочки (избыток шванновских клеток вокруг нервных волокон) и никаких признаков воспаления. Невролог заключил, что эти результаты сильно напоминают демиелинизирующую нейропа-тию, например болезнь Шарко-Мари-Тута 1-го типа, также известную как наследственная мотосенсорная нейропатия 1-го типа. Объяснив, что наиболее частая причина болезни Шарко-Мари-Тута — дупликация в гене периферического миелинового белка 22 (PMP22), невролог предложил анализ на эту дупликацию. Тест подтвердил, что пациентка имела удвоение аллеля PMP22 и болезнь Шарко-Мари-Тута 1А типа.
Читайте также:
