
Синдрома дефицита белка-транспортера глюкозы glut1 в цнс
Болезнь Де Виво характеризуется ранней эпилептической энцефалопатией, задержкой психомоторного развития, спастичностью, формированием микроцефалии, атаксией, дизартрией, альтернирующей гемиплегией, снижением уровня глюкозы и лактата в ликворе.
Эпилепсия при данном синдроме является фармакорезистентной и единственной терапией данного синдрома является кетогенная диета (до полной разработки генетической таргетной терапии).
Впервые синдром был описан в 1991 г профессором Де Виво (США)
На сегодняшний день различают две клинические формы:
- Классическая форма (примерно 80-90% больных): судороги начинаются в возрасте от 1 мес до 2 лет в 90%, после 2-х лет- у 10%
Отмечаются задержка психомоторного развития, дизартрия, атаксия, микроцефалия, дистония и хореатотоз, альтернирующая гемиплегия
- Неэпилептическая форма ( примерно 10% больных): мягкий фенотип без судорожного синдрома, но с выраженными дискинезиями, хореоатетозом, выраженной атаксией, альтернирующей гемиплегией,
- с пароксизмальной кинезиогенной дистонией
Атипичные формы (5%)
- Пациенты с абсансной эпилепсией с ранним началом (до 4 лет)
- Классическая генерализованная идиопатическая эпилепсия
- Детская абсансная эпилепсия
При синдроме дефицита GLUT1 в гене SLC2A1 обнаруживают мутации, которые обусловливают уменьшение транспорта глюкозы через клеточную мембрану. По данным исследований с использованием позитронно-эмиссионной томографии, регистрируется сниженный метаболизм глюкозы в базальных ганглиях. Глюкоза служит основным источником энергии для мозга, в том числе для процессов высвобождения нейротрансмиттеров, работы синапсов. Диффузия глюкозы через гематоэнцефалический барьер зависит от ряда переносчиков. Белок GLUT1 является ключевым, ответственным за доставку глюкозы через гематоэнцефалический барьер. Кодирующий GLUT1 ген SLC2A1 состоит из 10 экзонов и 9 интронов, локализующихся на коротком плече хромосомы 1 (1p34.2).
В настоящее время описано более 150 вариантов патогенных мутаций – точковые мутации, в том числе миссенс-мутации, нонсенс мутации, делеции/инсерции, варианты сайта сплайсинга, ведущие к сдвигу рамки считывания и инициации трансляции, а также крупномасштабные делеции и вариации числа копий.
В большинстве случаев заболевание возникает вследствие гетерозиготной мутации в гене SLC2A1, появляющейся преимущественно впервые (de novo). Реже происходит наследование от родителей по аутосомно-доминантному типу. Родитель, у которого обнаружена мутация SLC2A1, может иметь легкую форму заболевания. Причиной мягкого фенотипа может быть сниженная активность белкового продукта, а при тяжелых проявлениях - полная потеря функции. Реже возможно аутосомно-рецессивное наследование заболевания.
Мутации в гене SLC2A1 преимущественно связаны с генерализованными припадками, в большинстве случаев проявляющимися абсансными или миоклоническими приступами. Тот факт, что мутации SLC2A1 чаще встречаются у больных эпилепсиями с началом в раннем детском возрасте, включая абсансную эпилепсию с ранним дебютом и миоклонически-астатическую эпилепсию, говорит о возможных механизмах возрастной зависимости. Примерно 1 % генетических генерализованных эпилепсий ассоциированы с мутацией SLC2A1. И более 10% миоклонических абсансных эпилепсий и абсансных эпилепсий с ранним началом связаны с данной мутацией. Данные клинические картины вероятно связаны с возрастными изменениями созревания мозга и транспорта глюкозы, как показано с помощью позитронной томографии, и возрастной экспрессией гена SLC2A1.
Следовательно молекулярно-генетический анализ SLC2A1 оправдан у всех детей с абсансными приступами, особенно в случае начала припадков до 4-летнего возраста. Что является важным для раннего начала эффективного лечения с использованием кетогенной диеты и проведения генетического консультирования семьи.
Практически все пациенты имеют интеллектуальные нарушения разной степени тяжести, нарушения речи, дизартрию. Двигательные нарушения представлены атаксией, дистонией, хореей, данные нарушения могут быть как постоянными, так и пароксизмальными.
Электроэнцефалографическая картина при Глут 1 дефиците имеет также свои особенности. У детей раннего возраста часто регистрируется фокальное замедление или эпилептиформные разряды, а после 2-х лет – наиболее частая аномалия – генерализованные разряды пик-и поли-пик – медленная волна, частоты 2,5-4 Гц. Интересно наблюдение разницы между пре- и постпрандиальной ЭЭГ (до и после еды) – снижение индекса эпилептиформной активности после приема углеводов.
Пароксизмальный хореоатетоз, ранее известный как дистония со спастичностью, тип 9 (DYT9), а также дистония 18 типа рассматриваются как часть фенотипического спектра Глут1 дефицита.
Для диагностики синдрома необходимо проведение некоторых диагностических процедур: люмбальная пункция, которая позволит выявить низкий уровень глюкозы в спинно-мозговой жидкости (гипогликорахию), а также низкое соотношение глюкозы ликвора и крови
| Тип ГЛЮТ | Локализация в органах |
| ГЛЮТ-1 | преимущественно в плаценте, мозге, почках, толстой кишке, меньше в жировой ткани, мышцах |
| ГЛЮТ-2 | преимущественно в печени, β-клетках островков Лангерганса, энтероцитах |
| ГЛЮТ-3 | во многих тканях, включая мозг, плаценту, почки |
| ГЛЮТ-4 | инсулинозависимый, в мышцах (скелетных, сердечной), жировой ткани (находятся почти полностью в цитоплазме) |
| ГЛЮТ-5 | в тонкой кишке, в меньшей мере в почках, скелетных мышцах, жировой ткани, мозге. Переносчик фруктозы |
Все типы ГЛЮТ могут находиться как в плазматической мембране, так и в цитозольных везикулах. В клетках мышц и жировой ткани ГЛЮТ-4 (инсулинозависимые) почти полностью локализуются в цитоплазме. Влияние инсулина на такие клетки приводит к перемещению везикул, содержащих ГЛЮТ-4, к плазматической мембране и их слиянию с ней. После этого возможен облегченный транспорт глюкозы в клетки. При снижении концентрации инсулина в крови белки транспортеры глюкозы снова перемещаются в цитозоль и поступление глюкозы в эти ткани прекращается.
Гипо - и гипергликимии. Сахарный диабет.
В случае секреции слишком большого количества инсулина (например, при опухоли β-клеток - инсулиноме) или при передозировке экзогенно введенного инсулина, уровень глюкозы в крови падает, и может наступить гипогликемическая кома, которая может закончиться летально, если вовремя не будет оказана медицинская помощь.
Если инсулина вырабатывается слишком мало или он не может действовать на клетки-мишени, устанавливается высокий уровень сахара в крови (гипергликемия), и развивается сахарный диабет. Название этого заболевания Diabetus mellitus (diabetus — просеиваю, mellitus — сладкий) связано с тем, что моча становится сладкая на вкус, вследствие глюкозурии. При дефиците инсулина нарушаются все виды пластического, энергетического, водно-солевого обмена, страдают практически все функциональные системы.
Классификация сахарного диабета (1999 год)
1. Сахарный диабет типа I — деструкция ß-клеток, обычно приводящая к абсолютной инсулиновой недостаточности.
· Иммунно-опосредованный диабет (АДА) и аутоиммунный (ВОЗ)
2. Сахарный диабет типа II — от преимущественной резистентности с относительной или умеренной инсулиновой недостаточностью до преимущественного дефекта секреции инсулина с резистентностью к нему (АДА) или от преимущественной резистентности к инсулину с относительной инсулиновой недостаточностью до преимущественного секреторного дефекта с/или без инсулиновой резистентности (ВОЗ).
3. Другие специфические типы диабета:
· Генетические дефекты β-клеток (АДА),
· Генетические дефекты β-клеточной функции (ВОЗ)
· Генетические дефекты действия инсулина
· Болезни экзокринной части поджелудочной железы
· Диабет, индуцированный лекарствами или химическими веществами
· Диабет, вызванный инфекциями
· Необычные формы иммунно-опосредованного диабета
· Другие генетические синдромы, сочетающиеся с диабетом
4. Гестационный сахарный диабет (диабет беременных).
Инсулинзависимый сахарный диабет
Инсулинзависимый сахарный диабет (ИЗСД) — заболевание, вызываемое разрушением β -клеток островков Лангерганса поджелудочной железы.
Деструкция β-клеток — результат аутоиммунных реакций. В аутоиммунной реакции принимают участие лимфоциты и макрофаги (моноциты). Эти клетки продуцируют цитокины, которые либо непосредственно повреждают β-клетки, либо опосредуют клеточные реакции против β-клеток.
Провоцировать возникновение диабета I типа может вирусная инфекция, вызывающая деструкцию β-клеток. К таким вирусам, называемым β-цитотропными, относят вирусы оспы, краснухи, кори, цитомегаловирус, эпидемического паротита, Коксаки, аденовирус. Некоторые β-цитотропные вирусы вызывают лизис β-клеток.
Известны некоторые токсические вещества, например, производные нитрозомочевины и другие нитро- или аминосодержащие соединения, избирательно поражающие β-клетки и индуцирующие аутоиммунную реакцию. Кроме того, ИЗСД может быть результатом частичного генетически обусловленного дефекта системы иммунологического надзора и сочетаться с другими аутоиммунными заболеваниями. На долю ИЗСД приходится примерно 25-30% всех случаев сахарного диабета. Как правило, разрушение β-клеток происходит медленно, и начало заболевания не сопровождается нарушениями метаболизма. Когда погибает 80-95% клеток, возникает абсолютный дефицит инсулина, и развиваются тяжёлые метаболические нарушения. ИЗСД поражает в большинстве случаев детей, подростков и молодых людей, но может проявиться в любом возрасте (начиная с годовалого).
2. Инсулинонезависимый сахарный диабет
Инсулинонезависимый сахарный диабет (ИНСД) — общее название нескольких заболеваний, развивающихся в результате относительного дефицита инсулина, возникающего вследствие нарушения секреции инсулина, нарушения превращения проинсулина в инсулин, повышения скорости катаболизма инсулина, а также повреждения механизмов передачи инсулинового сигнала в клетки-мишени (например, дефекта рецептора инсулина, повреждения внутриклеточных посредников инсулинового сигнала и др.). ИНСД поражает людей, как правило, старше 40 лет. Сахарный диабет II типа характеризуется высокой частотой семейных форм. Риск ИНСД у ближайших родственников больного достигает 50%, тогда как при ИЗСД он не превышает 10%. Заболевание поражает преимущественно жителей развитых стран, особенно горожан.
Возможными причинами ИНСД могут быть: образование антител к рецепторам инсулина; генетический дефект пострецепторного аппарата инсулинзависимых тканей; нарушения регуляции секреции инсулина.
К факторам, определяющим развитие и клиническое течение болезни, относят ожирение, неправильный режим питания, малоподвижный образ жизни, стресс.
Мутации генов, контролирующих секрецию инсулина, энергетический обмен в β-клетках и обмен глюкозы в клетках-мишенях инсулина, приводят к возникновению нескольких форм ИНСД с аутосомно-доминантным наследованием.
Основным провоцирующим фактором инсулинонезависимого диабета служит ожирение.
Этот тип диабета часто сочетается с гиперинсулинемией, что способствует ожирению. Таким образом, ожирение, с одной стороны, важнейший фактор риска, а с другой — одно из ранних проявлений сахарного диабета.
При сахарном диабете, как правило, соотношение инсулин/глюкагон снижено. При этом ослабевает стимуляция процессов депонирования гликогена и жиров, и усиливается мобилизация запасов энергоносителей. Печень, мышцы и жировая ткань даже после приёма пищи функционируют в режиме постабсорбтивного состояния.
Симптомы сахарного диабета
Для всех форм диабета характерно повышение концентрации глюкозы в крови — гипергликемия.После приёма пищи концентрация глюкозы может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе.
Повышение концентрации глюкозы в плазме крови обусловлено снижением скорости использования глюкозы тканями вследствие недостатка инсулина или снижения биологического действия инсулина в тканях-мишенях.
При дефиците инсулина уменьшается количество белков-переносчиков глюкозы (ГЛЮТ-4) на мембранах инсулинзависимых клеток (жировой ткани и мышц). В мышцах и печени глюкоза не депонируется в виде гликогена, в жировой ткани уменьшается скорость синтеза и депонирования жиров. Кроме того, при снижении инсулинглюкагонового индекса активируется глюконеогенез из аминокислот, глицерола и лактата. Повышение концентрации глюкозы в крови при сахарном диабете превышает концентрационный почечный порог, что становится причиной выделения глюкозы с мочой (глюкозурия). В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу, если её уровень не превышает 8,9 ммоль/л (160 мг/дл).
К характерным признакам сахарного диабета относят также повышение концентрации в крови кетоновых тел - кетонемия.При низком соотношении инсулин/глюкагон жиры не депонируются, а ускоряется их катаболизм, так как гормончувствительная липаза в жировой ткани находится в фосфорилированной активной форме. Концентрация неэтерифицирован-ных жирных кислот в крови повышается. Печень захватывает жирные кислоты, окисляет их до ацетил-КоА, который, в свою очередь, превращается в β-гидроксимасляную и ацетоуксусную кислоты. В тканях ацетоацетат частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии. Увеличение концентрации кетоновых тел в крови (выше 20 мг/дл, иногда до 100 мг/дл) приводит к кетонурии. Накопление кетоновых тел снижает буферную ёмкость крови и вызывает ацидоз.
Ещё один характерный признак сахарного диабета — повышенный уровень в крови липопротеинов (в основном, ЛПОНП) — гиперлипопротеинемия. Пищевые жиры не депонируются в жировой ткани вследствие ослабления процессов запасания, а поступают в печень, где частично превращаются в триацилглицеролы, которые транспортируются из печени в составе ЛПОНП.
При сахарном диабете дефицит инсулина приводит к снижению скорости синтеза белков в организме и усилению распада белков. Это вызывает повышение концентрации аминокислот в крови. Аминокислоты поступают в печень и дезаминируются. Безазотистые остатки гликогенных аминокислот включаются в глюконеогенез, что ещё более усиливает гипергликемию. Образующийся при этом аммиак вступает в орнитиновый цикл, что приводит к увеличению концентрации мочевины в крови и, соответственно, в моче — азотемия и азотурия.
Высокие концентрации глюкозы, кетоновых тел, мочевины требуют усиленной экскреции их из организма. Поскольку концентрационная способность почек ограничена, резко увеличивается выделение большого количества воды, в результате чего может наступить обезвоживание организма. Выделение мочи у больных возрастает в несколько раз и в некоторых случаях достигает 8-9 л в сутки, но чаще не превышает 3-4 л — полиурия.Потеря воды вызывает постоянную жажду — полидипсия.
Нарушения углеводного обмена при сахарном диабете связаны с тем, что снижается поступление глюкозы в клетки из крови и все пути её использования (рисунки 3-4). Снижается гликолиз, окисление глюкозы, синтез гликогена, пентозофосфатный путь, синтез жиров из глюкозы. Наоборот, увеличивается мобилизация гликогена, глюконеогенез. Это вызывает гипергликемию – глюкозы много в крови и мало в тканях (“голод среди изобилия”). Из-за нарушения пентозо-фосфатного пути возникает дефицит НАДФН. Это способствует помутнению хрусталика (катаракта).
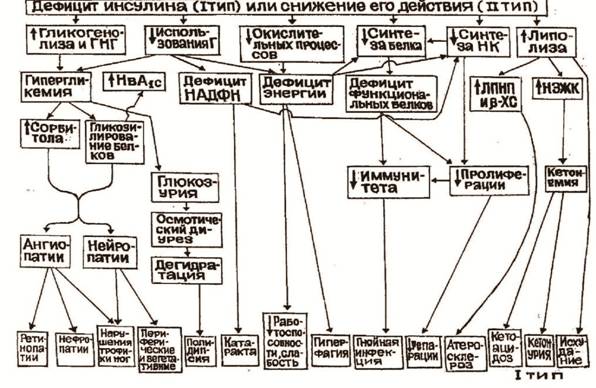
Рисунок 3 — Патогенез основных симптомов СД
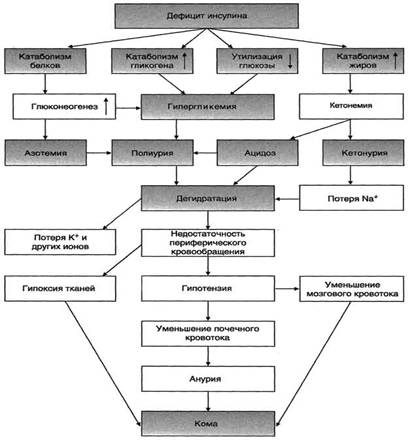
Рисунок 4— Изменение метаболизма при сахарном диабете
Глюкагон.
Глюкагон, образующийся в α-клетках островков Лангерганса представляет собой полипептид, состоящий из 29 аминокислотных остатков.
Синтез и секреция. Гормон синтезируется в α-клетках поджелудочной железы в виде неактивного предшественника препроглюкагона, который после отщепления N-конечной сигнальной последовательности превращается в проглюкагон и затем после воздействия протеаз - на глюкагон. В плазме глюкагон находится в свободной форме, поэтому период его полужизни составляет около 5 минут. Секреция глюкагона стимулируется аминокислотами, гастрином, катехоламинами и подавляется глюкозой, инсулином, жирными кислотами и Ca 2 + .
Механизм действия глюкагона.
Механизм действия глюкагона обусловлен его связыванием со специфическими глюкагоновыми рецепторами клеток печени (рисунок 5). Это приводит к повышению опосредованной G-белком активности аденилатциклазы и увеличению образования цАМФ. Результатом является усиление катаболизма депонированного в печени гликогена (гликогенолиза). Глюкагон для гепатоцитов служит внешним сигналом о необходимости выделения в кровь глюкозы за счёт распада гликогена (гликогенолиза) или синтеза глюкозы из других веществ — глюконеогенеза. Гормон связывается с рецептором на плазматической мембране и активирует при посредничестве G-белка аденилатциклазу, которая катализирует образование цАМФ из АТФ. Далее следует каскад реакций, приводящий в печени к активации гликогенфосфорилазы и ингибированию гликогенсинтазы. Этот механизм приводит к высвобождению из гликогена глюкозо-1-фосфата, который превращается в глюкозо-6-фосфат. Затем под влиянием глюкозо-6-фосфатазы образуется свободная глюкоза, способная выйти из клетки в кровь.
Также повышение содержания цАМФ индуцирует синтез ферментов глюконеогенеза в печени, усиливает превращение аминокислот в глюкозу. Таким образом, центральный эффект глюкагона — гипергликемия — обеспечивается двумя механизмами: быстрым (гликогенолиз) и медленным (глюконеогенез).
Глюкагон активирует гликогенолиз только в печени, в отличие от адреналина, который стимулирует этот процесс и в мышцах, и в печени. Глюкагон практически не оказывает действия на гликоген скелетных мышц, по-видимому, из-за практически полного отсутствия в них глюкагоновых рецепторов.

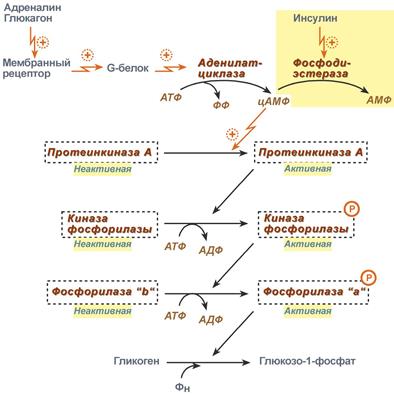
Рисунок 5 – Механизм действия глюкагона на клетку-мишень
Глюкагон вызывает увеличение секреции инсулина из здоровых β-клеток поджелудочной железы и торможение активности инсулиназы. Это является одним из физиологических механизмов противодействия вызываемой глюкагоном гипергликемии.
Таким образом биологическое действие глюкагона на углеводный обмен происходит в двух направлениях: активное участие в процессе гликогенолиза и стимуляция секреции инсулина.
Дата добавления: 2018-04-15 ; просмотров: 898 ;








Болезнь де Виво
В настоящее время проблема наследственных заболеваний занимает одно из ведущих мест в мире. С каждым годом увеличивается количество детей с различными врожденными пороками и аномалиями. К ним можно отнести болезнь де Виво ( синдром дефицита транспортера глюкозы 1 типа(ГЛЮТ-1)). Распространённость этой патологии на данный момент не известна.
ГЛЮТ обнаружены во всех тканях. Существует несколько разновидностей ГЛЮТ, они пронумерованы в соответствии с порядком их обнаружения.
ГЛЮТ-1 обеспечивает стабильный поток глюкозы в мозг;
ГЛЮТ-2 обнаружен в клетках органов, выделяющих глюкозу в кровь. Именно при участии ГЛЮТ-2 глюкоза переходит в кровь из энтероцитов и печени. ГЛЮТ-2 участвует в транспорте глюкозы в β-клетки поджелудочной железы;
ГЛЮТ-3 обладает большим, чем ГЛЮТ-1, сродством к глюкозе. Он также обеспечивает постоянный приток глюкозы к клеткам нервной и других тканей;
ГЛЮТ-4 - главный переносчик глюкозы в клетки мышц и жировой ткани;
ГЛЮТ-5 встречается, главным образом, в клетках тонкого кишечника. Его функции известны недостаточно.
Болезнь де Виво имеет аутосомно-доминантный тип наследования. Ген SLC 2 A 1, кодирующий белок ГЛЮТ-1, расположен на коротком плече 1 хромосомы и имеет в своем составе 10 экзонов и 9 интронов.
89% случаев патогенные варианты синдрома дефицита ГЛЮТ-1 представлены миссенс-, нонсенс-мутациями, вариантами сайта сплайсинга, сдвига рамки считывания и инициации трансляции. У остальных 11% изменениями в гене были внутригенные делеции и дупликации.
Причиной синдрома дефицита ГЛЮТ-1 является мутация в гене OMIM # 606777, ORPHA71277. Данное заболевание характеризуется эпилепсией, которая отвечает на кетогенную диету. Частичный дефицит глюкозного транспортера 1 типа ассоциирован с ограничением облегченной диффузии глюкозы через эндотелиальные и глиальные клетки капилляров головного мозга. Вследствие чего развиваются такие патологии как энцефалопатии, психомоторной задержкой со спастичностью, атаксией, дизартрией и альтенирующей гемиплегией, а также снижением глюкозы в ликворе.
Клинические проявления болезни де Виво провоцируются физической нагрузкой и голодом.
Пациенты, рожденные с данным синдромом, физиологически нормальной протекавшей беременности и родов, имеют нормальную массу тела.
Синдром дефицита глюкозного транспортера 1 типа представлен следующими формами:
Классическая форма ( самая распространённая,
90%). Данный тип болезни характеризуется задержкой психо-неврологической развития, дизартрией, формированием микроцефалии, а также двигательных расстройств. Судороги в основном начинаются от 1 месяца до 2 лет, но бывает и позже.
Неэпилептическая форма ( всего 10-15% больных). Пациенты имеют более мягкий фенотип без судорог, но с более выраженными пароксизмальными дискинезиями, в которую входят: дистония, реоатетоз, альтернативная гемиплагия.
Двигательные нарушения данного заболевания проявляются хореей, дистонией, атаксией. Длительные голодания, стресс, инфекционные заболевания являются вызывающими или усиливающими факторами проявления патологии.
При ранней классической форме болезни де Виво, уже в возрасте от 1 до 6 месяцев проявляются судорожные приступы ( клинический признак дисфункции мозга). Иногда судорожному синдрому предшествуют аномальные движения глазных яблок и апноэ. Описаны следующие типы судорог:
Частота приступов у пациента варьирует от ежедневных судорог до приступов, которые наблюдаются раз в несколько дней, недель, месяцев и не коррелируют с тяжестью фенотипа.
Помимо выше указанных дисфункций у больного наблюдается различная степень выраженности нарушения речи. Когнитивные нарушения представлены неспособностью ребенка к обучению, вплоть до тяжелой умственной отсталости, но социальное адаптивное поведение сохранено.
Профилактика и лечение наследственных заболеваний были и по-прежнему остаются одними из главных проблем общества. необъяснимые и спонтанные мутации значительно изменяют жизнь человека, в генотипе которого они содержатся.
Диагностика синдрома дефицита транспортёра глюкозы 1 типа основывается на клинической картине, биохимического анализа спинномозговой жидкости (уровень содержания глюкозы), медико-генетическое консультирование семьи, молекулярно-генетическое обнаружение мутаций в гене SLC2A.
Кетогенная диета является хорошо эффективной патогенной терапией, которая позволяет уменьшить клинические проявления: улучшить речь и движение, купировать судороги.
Болезнь де Виво связанная с нарушением транспорта глюкозы в головной мозг, приводит к неврологическим расстройствам с большим фенотипическим разнообразием и в настоящее время поддается эффективной терапии.
Список литературы
Биохимия: учебник/ под ред. Е. С. Северин изд. испр. и доп.:- М.: ГЭОТАР Медиа, 2013 - 768с.
• Для того чтобы преодолеть гематоэнцефалический барьер, глюкоза транспортируется в астроциты через эндотелиальные клетки небольших кровеносных сосудов
• Белки переносят глюкозу за счет унипорта, транспортируя ее в направлении градиента концентрации
• Белки, переносящие глюкозу, претерпевают конформационные изменения, которые приводят к реориентации их субстратных сайтов в мембране клетки
В клетках эукариот основным источником энергии является глюкоза, и многие клетки нуждаются в постоянном ее поступлении, поскольку у них глюкоза служит основным источником энергии для синтеза АТФ. Глюкоза представляет собой полярную молекулу, которая способна к гидратации, а клеточные мембраны не пропускают такие небольшие полярные метаболиты, как сахара. Поэтому для транспорта глюкозы в клетку требуется участие специфических мембранных белков. Транспорт глюкозы через плазматическую мембрану происходит с участием продуктов двух семейств генов.
Переносчики глюкозы (GLUTs) осуществляют независимый перенос (унипорт), при котором через мембрану происходит облегченный транспорт глюкозы. В противоположность GLUT-белкам, котранспортеры Na+/глюкоза расходуют энергию трансмембранного градиента Na+ на транспорт глюкозы. В настоящем разделе мы рассмотрим GLUT-белки.
![]()
Глюкоза селективно транспортируется через гематоэнцефалический барьер посредством изоформы 1 (GLUT-1) белка-переносчика.
Перенос глюкозы из крови в головной мозг и в другие отделы ЦНС представляет собой многоступенчатый процесс,
в котором участвуют различные типы клеток.
Семейство GLUT-белков является частью более обширного суперсемейства белков, облегчающих основные транспортные процессы (MFS) и широко представленных в клетках всех организмов. GLUT-белки представляют собой интегральные белки мембран клеток эукариот. Изоформы переносчиков GLUT различаются по своим кинетическим характеристикам, специфичностью по отношению к переносимым сахарам, тканевой локализацией и механизмами регуляции. Некоторые GLUT-белки, наряду с глюкозой, транспортируют и другие метаболиты, например галактозу, воду и анальгетики из группы гликопептидов.
GLUT-белки осуществляют унипорт, при котором вещества проходят через мембрану в направлении градиента концентрации. Таким образом, в зависимости от концентрации, GLUT-белки переносят метаболиты в клетку или в противоположном направлении. Поступление в клетку глюкозы, происходящее при участии GLUT-белков, часто определяет жизнеспособность клеток, которые характеризуются высоким уровнем потребления энергии.
Питательные метаболиты, такие как сахара, через кровеносные сосуды транспортируются в органы. Эндотелиальные клетки, выстилающие стенки небольших сосудов, контролируют процессы обмена питательных веществ. В этих эндотелиальных клетках, особенно расположенных в области гематоэнцефалического барьера, содержится много GLUT-белков. Надлежащее функционирование головного мозга сильно зависит от глюкозы, и его клетки особенно чувствительны к снижению ее содержания.
Транспорт глюкозы в нервные клетки происходит через капилляры мозга в несколько этапов и с участием изоформы GLUT-1. Эта изоформа экспрессируется в мембране клеток эндотелия, находящихся на границе между кровью и межклеточным пространством, а также в плазматической мембране астроцитов, функция которых важна в гематоэнцефалическом барьере. Расположенные в этих местах белки GLUT-1 транспортируют глюкозу из крови в эндотелиальные клетки, и оттуда в астроциты. В них глюкоза превращается в другие источники энергии, которые транспортируются в нейроны.
В различных тканях содержатся разные изоформы GLUT. Например, в клетках мышечной и жировой ткани транспорт глюкозы осуществляется с участием GLUT-4. В процессе приема пищи или после еды в клетки этих тканей под действием инсулина увеличивается поступление глюкозы. При этом происходит регулируемый транспорт изоформы GLUT-4 к клеточной поверхности. Эта изоформа также называется инсулин-зависимый переносчик. Белок GLUT-4 локализован во внутриклеточных везикулах, которые сливаются с плазматической мембраной. Этим обеспечивается доставка переносчика GLUT-4 к плазматической мембране и увеличивается емкость транспортного процесса.
![]()
Предполагаемое строение GLUT-переносчика, состоящего из 12 трансмембранных сегментов с внутриклеточными С- и N-концевыми участками.
Внутриклеточные петли содержат сайты фосфорилирования и связывания субстратов.
Показано, что градиент глюкозы по обеим сторонам мембраны может возникать в любом направлении, в зависимости от типа клеток и их метаболизма.
Градиент определяет направление транспорта.
Связывание инсулина со своим поверхностным рецептором запускает каскад внутриклеточных процессов, приводящих к быстрому слиянию этих везикул с мембраной. Это, в свою очередь, приводит к быстрому увеличению транспорта глюкозы в клетку с участием GLUT-4. При диабете типа II не происходит транспорт глюкозы из крови в мышцы и жировую ткань, очевидно, из-за нарушения позиционирования GLUT-4 на плазматической мембране. Более того, GLUT-2 способен экспортировать глюкозу из клеток тех органов, в которых она образуется, например из клеток печени.
По строению переносчик GLUT напоминает другие члены суперсемейства MFS. Предполагается, что он состоит из 12 трансмембранных а-спиралей, имеющих внутриклеточные N- и С-концевые участки и петли. Последние содержат сайт связывания субстрата и сайты фосфорилирования.
Модель структуры GLUT-1 предложена на основании результатов по сайт-направленному мутагенезу и по измерению транспорта глюкозы мутантным белком. Для построения модели также использовались данные по кристаллической структуре бактериальной лактопермеазы, относящейся к подсемейству переносчиков олигосахари-дов/Н+, входящих в MFS.
Согласно этой модели, при соответствующей ориентации трансмембранных спиралей создается полость поры, через которую транспортируется глюкоза, а также обеспечивается образование водородных связей между GLUT-белком и глюкозой. Результаты кинетического анализа транспорта глюкозы в эритроцитах позволяют предполагать существование специального механизма, который обеспечивает белку GLUT возможность принимать одну из двух основных конформаций. Этот механизм аналогичен постулируемому для бактериальной лактопермеазы. Когда белок находится в одной конформации, сайт связывания глюкозы обращен в сторону внеклеточного пространства. В другой конформации он обращен в сторону цитозоля. При связывании глюкозы с любой стороны происходят конформационные изменения, которые приводят к реориентации сайтов связывания глюкозы к противоположной стороне мембраны и к ее высвобождению.
Таким образом, хотя переносчики GLUT являются унипортерами, а бактериальная лактопермеаза представляет собой симпортер, предполагается, что они функционируют по одному механизму.
Мутации в гене GLUT-1 вызывают значительные дефекты развития. У детей потребность головного мозга в глюкозе в 3-4 раза выше, чем у взрослых, и в мозг поступает до 80% всей глюкозы. Мутации в гене GLUT-1 человека служат причиной проявления редкого синдрома недостаточности GLUT-1, который характеризуется развитием припадков и замедлением развития. Считается, что это связано с нарушением транспорта глюкозы в головной мозг. Эмбрионы мышей, дефектных по гену GLUT-1, отстают в росте, и у них обнаруживаются различные пороки развития. Аналогичные дефекты возникают у мышиных эмбрионов, которые развиваются в организме диабетических самок. При этом избыток глюкозы в крови подавляет экспрессию GLUT-1 в органах эмбриона.
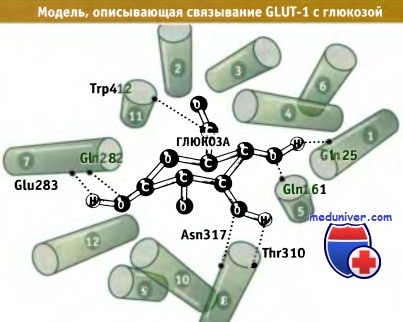
Предполагаемая ориентация трансмембранных а-спиралей переносчика глюкозы GLUT-1.
Показаны остатки, участвующие в связывании глюкозы (размер молекулы глюкозы представлен без учета масштаба).
Вид на спирали со стороны внутренней части плазматической мембраны.
Эта модель построена с использованием гомологичной структуры лактопермеазы E.coli в качестве матрицы.
Читайте также:
