
Эпилепсия и аномалия арнольда киари 1

К редким наследственным болезням относят аномалию Арнольда-Киари. Патологию назвали в честь врачей, которые классифицировали порок мозга в 19 и 20 веке с разницей в 100 лет. Выделяют 4 типа дефекта. Его диагностируют на МРТ, в лечении используют консервативные и хирургические методы.
- Что это такое?
- Причины
- 1 тип заболевания
- 2 тип
- 3 тип
- 4 тип
- Симптомы и признаки аномалии Арнольда-Киари
- Признаки 1 типа аномалии
- Признаки 2 степени
- Признаки 3 степени
- Признаки 4 типа
- Диагностика
- Лечение аномалии Арнольда-Киари
- Прогноз на жизнь
- Что нужно запомнить?
Что это такое?
Аномалией (синдромом, мальформацией) Арнольда-Киари называют порок краниовертебрального перехода. Он характерен смещением нижних мозговых структур к отверстию возле верхнего отдела хребта.
То есть, мозжечковые миндалины со стволом головного мозга выжимаются к позвоночному каналу (на уровне 1―2 позвонка) из задней ямки. Эта внутричерепная область находится между затылочной, клиновидной и височными костями над большим отверстием (здесь начинается хребет).

Наглядная демонстрация заболевания
Особенности аномалии Арнольда-Киари:
- есть 4 типа дефекта (степени тяжести);
- с жизнью совместима 1-2 степень;
- низ мозжечка смещена вниз с места нормы размещения;
- мозговые структуры не вмещаются в черепной задней ямке;
- мозжечком сдавливается продолговатый мозг, ликворные пути;
- нарушена циркуляция крови, спинномозговой жидкости;
- провоцирует развитие болезней ЦНС;
- симптомы могут возникнуть после совершеннолетия;
- лечится хирургическими методами;
- до изобретения МРТ трудно диагностировалась.
Аномалия причислена к порокам развития мозговых и черепных структур. Раньше считалась врожденной патологией. Сейчас врачи склоняются к мнению, что мальформация возникает из-за несоразмерного роста затылочной кости и какой-либо части головного мозга. Дефект может образоваться во внутриутробный период, у детей любого возраста, пока растет скелет.
Согласно статистике, дефект Арнольда-Киари выявляют максимум у 9 со 100 000 населения. Опасность аномалии заключается в развитии неврологического дефицита: у пациента нарушаются функции ЦНС. При несвоевременном выявлении либо неправильном лечении порок становится причиной инвалидности, смерти. Летальный исход чаще наступает из-за поражения дыхательного центра.
Причины
Выделяют три анатомические причины порока:
- Размер черепной задней ямки меньше нормы.
- Головной мозг либо мозжечок быстрее увеличивается, чем растет череп.
- Недостаточно развит фиксирующий аппарат краниовертебрального перехода (связки вверху хребта).
Во всех случаях мозговая ткань выжимается в затылочное отверстие, к 1 шейному позвонку.
Патологические причины аномалии Арнольда-Киари:
- краснуха, прочие опасные инфекции во время беременности;
- прием лекарств с тератогенным и эмбриотоксическим действием;
- родовая травма черепа;
- осложнение родов — применение вакуума, щипцов, отказ от кесарева при узком тазе;
- черепно-мозговая травма — ДТП, нападение, иные несчастные случаи;
- интракраниальные или внутричерепные опухоли;
- водянка, иные болезни мозга, вызывающие смещение структур вниз;
- генетические расстройства.
Справка! Среди причин синдрома Арнольда-Киари называют алкогольную/наркотическую зависимость будущих отца и/или матери, которая появилась еще до зачатия ребенка.
Во время беременности к провоцирующим факторам аномалии ребенка относят вредные привычки матери, пассивное курение — пребывание в прокуренных местах, рядом с курящими. Вызвать нарушение развития может влияние радиации, канцерогены, действие токсических веществ.
1 тип заболевания
При 1 степени мальформации смещаются вниз мозжечковые 1―2 миндалины в позвоночный канал. Нет острого нарушения циркуляции ликвора, ухудшения кровоснабжения мозга, неврологического дефицита. Считается наименее опасным для жизни, редко вызывает полную инвалидность. Чаще выявляют случайно во время обследования. Первые симптомы могут появиться после 30 лет либо никогда не возникнуть.
2 тип
При второй или детской степени мальформации удлиняется ствол головного мозга. Дисплазированный (аномально развитый) мозжечок вклинивается в затылочное отверстие. Патология Арнольда-Киари проявляется с рождения бульбарным синдромом. Характерно шумное дыхание с периодической остановкой (апноэ), нарушение глотательной функции.
Ребенок часто срыгивает, давится молоком, пища попадает в нос. К особенностям второго типа относят наличие сопутствующих врожденных патологий. Болезнь нередко сопровождается:
- спинномозговой грыжей (менингоцеле) в пояснице;
- черепно-мозговой грыжей (менингоэнцефалоцеле);
- стенозом (сужением) ликворных путей;
- сирингомиелией (кистой в спинном мозге);
- гидроцефалией.
Важно! Сопутствующие пороки требуют экстренной операции в период новорожденности. Хирургическое лечение повышает выживаемость малышей, снижает вероятность паралича, смерти.
3 тип
При 3 степени вниз полости смещаются и ущемляются ткани мозжечка, мозгового ствола. Структуры чаще выявляют вне черепной ямки в грыжевом мешке. Аномалии характерно превышение размера большого отверстия, дефект нервной ткани, затылочной кости, позвонков. Всегда сопровождается нарушением циркуляции ликвора, дисфункцией сердечно-сосудистой системы, дыхательного центра.
Третий тип врачи считают самым опасным среди разновидностей порока, в 95% случаев заканчивается смертью. Если ребенок выживает, за ним нужен пожизненный уход по причине инвалидности.
4 тип
Четвертую степень мальформации Арнольда-Киари часто называют аномалией Денди-Уокера. Проявляется с рождения младенца. Особенность порока заключается в недоразвитости и дисфункции мозжечка без его смещения вниз. Сопровождается сужением/отсутствием отверстий для оттока ликвора. Болезни характерно развитие умственной и физической недееспособности.
У детей болезнь 4 типа часто заканчивается смертью в первые три месяца после рождения, несмотря на своевременность лечения. В случае выживания за ребенком нужен беспрерывный уход, врачебное наблюдение. Инвалидность 1―3 группы оформляют сразу после постановки диагноза.
Симптомы и признаки аномалии Арнольда-Киари
Болезнь сопровождается специфическими синдромами. Выраженность и количество этих симптомокомплексов зависит от тяжести порока, какие внутричерепные и позвоночные структуры затронуты патологией.
Характеристика неврологических синдромов:
Тремор (дрожание) пальцев, ухудшение речи, двоение картинки (нарушение зрения).
Тошнота, рвота вне зависимости от приема лекарств, пищи, напряжение мышц на затылке.
Боль/дискомфорт в зоне затылка. Усиливается во время чихания, движений головы.
Онемение участков кожи, тела.
Ухудшение мышечной силы.
Слабость и атрофия мышц конечностей, туловища, головы, деформация мелких суставов, искривление позвоночника.
Справка! Аномалии Арнольда-Киари характерно как бессимптомное течение, так и медленное развитие, бурное прогрессирование. Признаки могут проявляться с рождения либо отсутствовать всю жизнь.
В случае симптомного течения болезни у человека наблюдается до трех синдромов: вертебробазилярной недостаточности, гипертензионно-гидроцефальный и/или мозжечковый. При этой тяжести порока Арнольда-Киари возможны регулярные приступы мигрени, распирающая боль в зоне затылка. Ухудшается четкость движений, мелкой моторики.
Наблюдаются признаки скачков давления и гипоксии:
Человек ощущает дискомфорт сзади у основания черепа, при поворотах/наклонах головы. В случае прогрессирования возможно присоединение других симптомокомплексов. Это проявление бульбарно-пирамидного, корешкового и/или сирингомиелического синдрома.
При втором типе порока врачи выявляют одновременно минимум три неврологических синдрома. Родившийся ребенок мало двигается, слабо кричит, ручки согнуты, мышцы напряжены. Есть дисфункция нервной системы, органов.
Пороку Арнольда-Киари 2 типа характерно:
- систематическая остановка дыхания;
- ослабление глоточных мышц;
- синюшность кожи.
Осложняется вторая степень частичным либо полным параличом. Утрата двигательных функций отмечается в пальцах, руке/ноге, в области головы, всего тела.
Третьей степени мальформации Арнольда-Киари характерно проявление 5―6 синдромов одновременно. Ребенок не кричит сразу после рождения. У пациентов часто отмечается потеря слуха, зрения, дисфункция выделительных органов. Нет четкой координации движения, по утрам возникает рвота, есть аномалии строения позвоночника, головы.
У людей с четвертой степенью тяжести порока преобладает мозжечковый и гипертензионно-гидроцефальный синдром. Дети беспокойны, слабо вскрикивают, тихо плачут. Если выживают, на обследованиях выявляют снижение интеллекта, отсутствие нормального физического развития. В целом, симптомы с прогнозом жизни третьей и четвертой степени тяжести похожи.
Диагностика
Для обследования обращаются к неврологу. Врач пальпирует шею, голову, изучает симптоматику, тяжесть сопутствующих синдромов, семейный и индивидуальный (пациента) анамнез, течение беременности. В ходе диагностики дополнительно консультируются с нейрохирургом.
Подтверждается порок краниовертебрального перехода только МРТ. МСКТ, компьютерная томография малоинформативны без применения контраста. Рентген и УЗИ не выявляют смещения мозговых структур. Ультразвуковая диагностика помогает выявить во время беременности у ребенка внутри утробы признаки гидроцефалии, неправильной формы черепа, мозжечка.
Важно! Поскольку мальформация часто сочетается с сирингомиелической кистой, томографией рекомендуют обследовать позвоночник.
Лечение аномалии Арнольда-Киари
Людей с бессимптомным пороком Арнольда-Киари ежегодно обследуют, чтобы своевременно выявить начало прогрессирования, осложнения. Пациентов с проявлением синдромов лечат лекарствами и методами физиотерапии. Показаны лечебные ванны, массаж, ЛФК. В случае выраженных нарушений циркуляции ликвора, риска смерти делают операцию.
Медикаментозная терапия болезни Арнольда-Киари:
| Группа лекарств | Действие | Цель применения |
| Нестероидные противовоспалительные средства | Предупреждают и лечат воспаление тканей | Устраняют болевой синдром |
| Ненаркотические анальгетики + спазмолитики, препараты нейромышечной блокады | Обезболивают | |
| Миорелаксанты | Снимают мышечное напряжение | |
| Мочегонные, прочие лекарства дегидратационной терапии | Выводят жидкость, снижают давление | Частично снимают симптомы, вызванные скоплением ликвора/крови в основании черепа |
| Нейропротекторы | Предупреждают повреждение, гибель нервных клеток | Убирают кислородное голодание тканей, улучшают регенерацию и функционирование ЦНС, мозга |
| Метаболики | Нормализуют обменные процессы | |
| Витамины группы В | Противосудорожные, восполняют дефицит веществ, улучшают регенерацию клеток | |
| Антиоксиданты | Подавляют окисление, образование радикалов | |
| Вазодилататоры | Сосудорасширяющие | |
| Ангиопротекторы | Укрепляют сосудистые стенки |
Операция показана людям с выраженным нарушением циркуляции ликвора, расстройством двигательной способности, усугублением болезни. Хирургическое лечение проводят при неэффективности лекарственной терапии в течение 60―90 дней, риске инвалидности, смерти.
Виды операций при мальформации Арнольда-Киари:
Справка! Операцией устраняют препятствие к движению ликвора, увеличивают пространство для смещенных структур мозга, снимают давление на нервы. Хирургическое лечение утраченные функции ЦНС не восстанавливает.
Прогноз на жизнь
Своевременность диагностики, вид мальформации и правильность лечения влияют на прогноз жизни. Быстрое выявление и начало терапии дефекта Арнольда-Киари I―II степени дают хорошие результаты. У подавляющего большинства прооперированных людей убираются проявления синдромов, у некоторых полностью исчезает неврологический дефицит. Несвоевременное лечение осложняется инвалидностью.
При третьем типе мальформации прогноз плохой. Много пациентов умирает, несмотря на своевременно сделанную операцию. Порок IV степени чаще оканчивается инвалидностью. Выживаемость и сохранение двигательной способности повышается в случаях смещения мозговых тканей в пределах черепной ямки над позвоночным каналом.
Эффективность хирургического лечения статистика подтверждает минимум у 50% пациентов, а отсутствие результата – у 15%. После операции бывают рецидивы синдрома Арнольда-Киари в первые три года.
Посмотрите видео по теме статьи. Нейрохирург А. А. Реутов рассказывает о своём видении проблемы аномалии Арнольда-Киари.
Что нужно запомнить?
- Аномалию Арнольда-Киари относят к наследственным патологиям с непредсказуемым течением.
- Среди причин указаны пороки развития мозга и черепа, травмы, инфекции.
- Первый тип чаще не влияет на продолжительность жизни, трудоспособность.
- Вторая степень тяжести требует своевременного проведения операции.
- Наиболее опасной аномалией считается 3 тип тяжести.
- Аномалия 4 степени по современной классификации больше подходит к аномалии Денди-Уокера, но также опасна для жизни.
- Благоприятный прогноз жизни врачи дают лишь пациентам с 1 типом аномалии Арнольда-Киари.
Список литературы
- Unal M, Bagdatoglu C. Arnold-Chiari type I malformation presenting as benign paroxysmal positional vertigo in an adult patient. J Laryngol Otol. 2007 Mar;121(3):296-8. Epub 2006 Dec 14.
- Milhorat TM, Nishikawa M, Kula RW, et al. Mechanisms of cerebellar tonsil herniation in patients with Chiari malformations as a guide to clinical management. Acta Neurochir (Wien). 2010 Jul;152(7):1117-27. doi: 10.1007/s00701-010-0636-3. Epub 2010 May 4.
- Федирко ВА. Синдромы нейроваскуляр-ной компрессии образований задней черепной ямки в сочетании с аномалией Киари. Украинский нейрохирургический журнал.
- Cirignotta F, Coccagna G, Zucconi M, et al. Sleep apneas, convulsive syncopes and auto-nomic impairment in type I Arnold-Chiari malformation. Eur Neurol. 1991;31(1):36-40.
- Speer MC, Enterline DS, Mehltretter L, et al. Chiari type I malformation with or without syringomyelia: prevalence and genetics.
В последнем десятилетии XIX века немецкий патологоанатом Киари описал четыре врожденные аномалии мозжечка и ствола мозга, как проявления нарастающей тяжести единого патогенетического механизма, связанного с давлением сзади на задний мозг при врожденной гидроцефалии.
Основной анатомической особенностью аномалии Киари I типа является разная степень каудального смещения миндалин мозжечка в шейный канал. Аномалия Киари 2 типа характеризуется более тяжелой мозжечковой грыжей, которая, кроме миндалин мозжечка, также включает в себя нижний червь и четвертый желудочек; она, как правило, связана с миеломенингоцеле. Аномалия Киари 3 типа включает в себя признаки высокого менингоэнцефалоцеле с грыжей заднего мозга. Наконец, Киари 4 типа характеризуется тяжелой гипоплазией мозжечка без каудального смещения.
После многих лет и большого количества теоретических, клинических и нейровизуализационных исследований, понимание и осознание этого состояния вышло на новый уровень. В настоящее время считается, что эти четыре порока развития являются отдельными состояниями, с различным патогенезом, клиническим проявлением, и прогнозом, а не разными прогрессирующими стадиями одного заболевания. С анатомической точки зрения аномалии 1-3 типа являются грыжами заднего мозга различной степени, с возможным вторичным образованием полости в спинном мозге, в то время как аномалия 4 типа характеризуется гипоплазией мозжечка. Гипоплазия задней черепной ямки является частой находкой при первых трех аномалиях, но никогда не встречается при четвертой.
У каждой формы свои патогномоничные клинические проявления и по этим причинам различные аномалии Киари должны рассматриваться в разных разделах.
Мальформация Киари 1 типа. Аномалия Киари 1 типа характеризуется неправильным положением (и формой) миндалин мозжечка, которые опускаются из полости черепа ниже большого затылочного отверстия в шейный канал. Это анатомическое нарушение приводит к окклюзии субарахноидального пространства на уровне большого затылочного отверстия с последующим нарушением ликвороциркуляции в головном и спинном мозге; фактически, помимо изменений в задней черепной ямке, повышение ликворного давления на этом уровне приводит к сирингомиелии. Патогенетические механизмы развития аномалии детально изучены и обсуждены в литературе. Различные механизмы патогенеза можно схематично разделить на три категории:
- Гидродинамический (на основе градиента давления между ликворными пространствами головного и спинного мозга).
- Механический (с блоком ликвороциркуляции на уровне большого затылочного отверстия как причины аномалии Киари 1 типа).
- Нарушение развития (с интерпретацией аномалии задней черепной ямки как местного проявления более общего нарушения развития).
Хотя аномалия Киари 1 типа обычно становится симптоматичной в раннем подростковом периоде, в последнее время она обнаруживается с одинаковой частотой как у детей, так и у взрослых.
а) Клиническая картина аномалии Киари I типа. Наиболее частой жалобой у этой группы пациентов является боль в затылке или в шее, усиливающаяся при чихании, кашле или при пробе Вальсальвы. Другими болезненными проявлениями являются боль в плече, спине или боли в конечностях без корешкового распределения. Наиболее частым симптомом являются признаки моторного или сенсорного дефицита в конечностях (> 70%), которые являются проявлением наличия полости в спинном мозге (сирингомиелии). Атаксия тела и конечностей в качестве проявление нарушений в мозжечке является вторым наиболее распространенным симптомом (в 30-40%). Реже (в 15-25%) наблюдается неуклюжесть, нистагм, диплопия, дисфагия и дизартрия как проявление дефицита черепных нервов. Апноэ при заикании наблюдается в 10% случаев, в большинстве случаев проявляясь у младенцев или маленьких детей. Своеобразным проявлением аномалии Киари 1 типа у детей и подростков является прогрессирующий сколиоз (в 30%).
б) Лучевая диагностика. МРТ — лучший способ для диагностики аномалии Киари 1 типа. Важными критериями для этого порока являются: пролабирование одного или обоих миндалин мозжечка ниже большого затылочного отверстия (> 5 мм); возможны шейно-мозговая деформация, отсутствие супратенториальных нарушений (за исключением отдельных случаев небольшого расширения желудочков и обычная локализация четвертого желудочка. Аномалия Киари 1 типа может быть связана с такими костными аномалиями, как маленькая задняя черепная ямка, платибазия, атланто-затылочная ассимиляция, базилярная импрессия, срастание шейных позвонков (аномалия Клиппеля-Фейля). Гидро/сирингомиелия встречается в 50-60% случаев. Она может быть ограничена одной или двумя областями, а может распространяться на всю длину спинного мозга. Обычно полость образуется на уровне С1.
МРТ является полезным дополнением к диагностическим мероприятиям, так как позволяет определить степень сдавления ствола на уровне затылочного отверстия и характеристики ликвородинамики; исследование, выполненное в послеоперационном периоде, позволяет получить некоторое представление об адекватности хирургической декомпрессии.
в) Лечение аномалии Киари I типа. Целью хирургического лечения является декомпрессия задней черепной ямки для восстановления циркуляции спинномозговой жидкости в базальных цистернах и устранение сдавления нервных структур на уровне краниоцервикального перехода. Хирургическое лечение при аномалии Киари 1 типа обычно определяет четко определенный протокол, который включает субокципитальную краниотомию, С1 ламинэктомию, лизис арахноидальных сращений, резекцию миндалин мозжечка (как традиционную тонзилэктомию, так и субпиальную коагуляцию) и расширенную дуропластику. В случае сопутствующей вентральной компрессии (как, например, при платибазии, С1 ассимиляции и т.д.), она должна быть устранена до проведения дорсальной декомпрессии. В последнее время появляются сообщения, которые указывают на возможность достижения сопоставимых результатов при использовании простой декомпрессии, без расширяющейся дуропластики (или с отслоением наружного слоя твердой мозговой оболочки). Интраоперационная ультразвуковая диагностика может быть полезна в этом отношении, демонстрируя синхронизацию движения миндалин с дыханием и сердцебиением, а также наличие адекватного движения ликвора из четвертого желудочка.
Дальнейшие хирургические возможности представлены процедурами, направленными на лечение гидро/сирингомиелии (закупорка задвижки, размещение сиринго-субарахноидального или сиринго-плеврального шунта, и стентирование четвертого желудочка). При сравнении двух основных крупных хирургических вмешательств при лечении аномалии Киари 1 типа с сирингомелией — субокципитальной декомпрессии и установки сиринго-субарахноидального шунта—Hida et al. обнаружили уменьшение размера сирингомиелитической полости у 94% пациентов, перенесших декомпрессию, и у 100% перенесших сирингосубарахноидальное шунтирование. Что касается прогноза, то максимальные преимущества декомпрессии проявляются у пациентов с симптомами, связанными с пароксизмальной внутричерепной гипертензией. Кроме того, пациенты с мозжечковой симптоматикой и синдромом большого затылочного отверстия имеют больше шансов на выздоровление, чем пациенты с центральным синдромом спинного мозга.
При работе с пациентами детского возраста главной проблемой являются показания к операции. Фактически, во многих случаях диагноз аномалии Киари ставится случайно, после МРТ для диагностики причины неспецифических клинических проявлений, таких как головные боли, умственная отсталость, эпилепсия. Большинство авторов не согласно с превентивным хирургическим лечением, считая, что показания к нему должны основываться на наличии симптомов и клинических проявлений, однозначно относящихся к аномалии Киари, а не данными нейровизуализации.
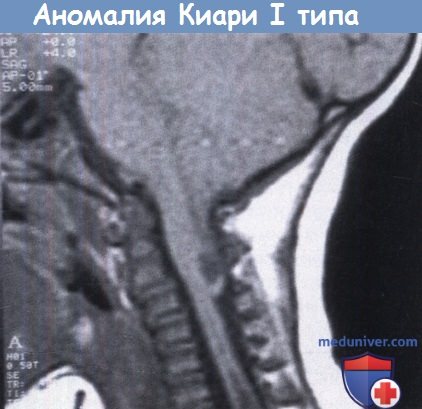
Аномалия Киари 1 типа.
Сагиттальный срез в Т1-взвешенном режиме МРТ.
Общие сведения
Синдром Арнольда-Киари является пороком развития мозжечка – отдела головного мозга, отвечающего за координацию, мышечный тонус и равновесие. Патологии присвоен код по МКБ-10 Q07.0 и она представляет собой опущение миндалин мозжечка вниз на уровень первого, а порой второго шейного позвонка (ниже черты Чемберлена) и блокирует нормальный ток спинномозговой жидкости.
Заболевание чаще всего сочетается с микрогирией, сдавливанием заднего отдела мозга, стенозом водопровода мозга, базилярной импрессией, инвагинацией, недоразвитием четверохолмия и другими мальформациями нервной системы. Синдром чаще всего встречается у особ в возрасте 12-71 год и не превышает 000,9%.
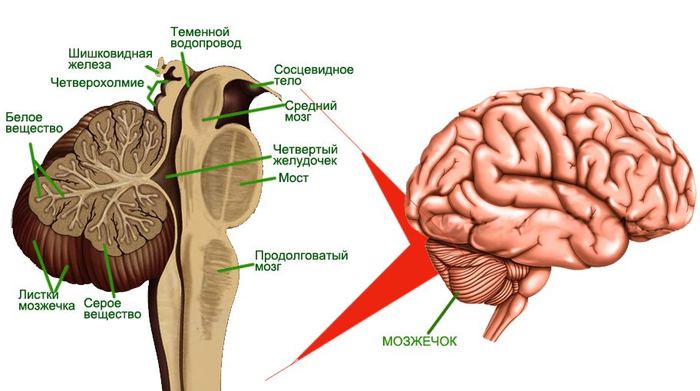
Локализация и строение мозжечка
Патогенез
В основе патофизиологии обычно лежит несоответствие размеров задней черепной ямки и имеющихся в ней структур нервной системы, а также:
- развитие аномалий тел шейных позвонков, включая их расщепление чаще всего первого (данный механизм развития встречается в 5% случаев), ассимиляцию атланта — сращивание шейного позвонка с затылочной костью;
- смещение структур мозжечка в период бурного роста мозга при медленно растущих костях черепа;
- гидроцефалия – избыточное скопление цереброспинальной жидкости;
- сирингомиелия – анормальный процесс развития полостей в спинном мозге;
- миеломенингоцеле – врожденный дефект развития нервной трубки;
- различные врожденные заболевания, в том числе платибазия, аномалия Денди-Уокера.
Классификация
В зависимости от клинической картины и степени развития анатомических аномалий синдром Арнольда-Киари бывает четырех типов.
Синдром Арнольда-Киари 1 типа проявляется в виде проникновения миндалин мозжечка в полость позвоночного канала, вызывающего гидромиелию и опущение структур задней черепной ямки ниже большого затылочного отверстия на 3-5 мм и более, причем нет никаких других мальформаций нервной системы. Средняя продолжительность жизни обычно не превышает 25-40 лет.
Представляет собой опущение в полость позвоночного канала различных структур мозжечка и тканей ствола, при этом данная нейропозвоночная мальформация сочетается с миеломенингоцеле (врожденной спинномозговой грыжей) и гидроцефалией. Манифестация происходит практические сразу после рождения.
Мальформация отличается наличием затылочного энцефалоцеле и различных признаков аномалии второго типа. Обычно не совместима с жизнью.
По своей сути это аплазия либо гипоплазия всех или отдельных структур мозжечка, то есть бывает тотальной и субтотальной. Первый вариант встречается достаточно редко и сочетается с прочими тяжёлыми аномалиями и заболеваниями нервной системы, включая анэнцефалию, амиелию. При субтотальной агенезии наблюдаются пороки развития других участков головного мозга, например агенезия моста, отсутствие четвёртого желудочка и пр.
Гипоплазия мозжечка встречается в форме уменьшения всего мозжечка или охватывает отдельные части, при этом сохраняются нормальные структуры без утраты функций. Встречается одно- и двусторонняя, лобарная, лобулярная и интракортикальная гипоплазия. Изменения конфигурации листков мозжечка обычно представлено в виде аллогирии, полигирии или агирии.
Кроме того, некоторые авторы выделяют два дополнительных типа:
Причины
Помимо роли наследственного и генетического фактора существует несколько теорий возникновения пороков мозжечка. Традиционная теория говорит, что опущение миндалин вызвано натяжением струны спинного мозга в результате напряжения концевой нити при развитии той или иной мальформации. Исключением становится болезнь Киари 1 типа, ведь единственным нарушением в этом случае становится опущение миндалин и оно может быть спровоцировано:
- гидродинамическими явлениями — нарушением циркуляции спинно-мозговых жидкостей;
- черепно-мозговыми и родовыми травмами;
- мальформацией — маленькие размеры и ограниченность затылочного отверстия могут приводить к опущению миндалин в просвет позвоночного канала;
- анормально натянутой связкой — так называемой концевой нитью (по теории доктора М.Б. Ройо Сальвадора — Filum System).
Кроме того, ученые выделяют ряд факторов, которые могут повысить риск развития аномалии Арнольда-Киари первого типа:
- генетическая предрасположенность – наличие патологии у родителей и более дальних предков, хотя хромосомных аномалий до сих пор не выявлено;
- травмы, особенно падения, могут вызвать компрессию и усиление натяжения концевой нити и привести к опущению миндалин мозжечка;
- неправильное поведение и вредные привычки женщины в период вынашивания младенца — злоупотребление и хаотичный прием медикаментов, курение, употребление алкоголя, а также перенесенные вирусные заболевания.
Симптомы
Симптоматика при различных типах синдрома Арнольда Киари может существенно отличаться – все зависит от степени натяжения и компрессии нервных структур затылочного отдела, но чаще всего у больных наблюдаются:
- головные боли;
- прогрессирующее увеличение размеров окружности головы;
- периодические боли в различных отделах позвоночника;
- парез и боль в различных областях конечностей;
- нарушения зрения и чувствительности, включая дизестезии и парестезии;
- шумы в ушах;
- паралич лицевого, глазодвигательного нерва;
- дрожь;
- приступы головокружения;
- бессонница;
- рвота;
- обмороки;
- нистагм;
- апноэ;
- нарушение работы сфинктеров, мышц языка, глотки, онемение и различные проявления мышечной слабости;
- нарушения способности глотания (дисфагия);
- нарушения памяти;
- несогласованность движений (атаксия) и как следствие — неуклюжая походка, причем нарушения наиболее ярко выражены на этапе формирования;
- сколиоз;
- трудности с удержанием позиций тела и равновесия, а также координации;
- затруднения при желании выразить мысль и подобрать слова.
Проявления синдрома Арнольда-Киари хронические, имеют тенденцию увеличивать интенсивность, что с каждым разом существенно ухудшает состояние больного и ограничивает его привычный образ жизни.
Самое опасное, что аномалия Арнольда-Киари может привести к внезапной смерти, ведь спинно-мозговые центры отвечают за сердечно-дыхательные функции, а давление миндалин мозжечка на них может спровоцировать остановку дыхания — апноэ, которое станет причиной летального исхода.
Патология 1 степени обычно выявляется случайно во время проведения МРТ, ведь больные помимо эпизодов апноэ и обмороков могут испытывать только:
- боль в шейно-затылочном отделе;
- снижение чувствительности.
Анализы и диагностика
При первых признаках необходимо пройти неврологический осмотр и провести оценку выраженности клинико-функциональных нарушений. Для подтверждения диагноза чаще всего применяются:
- нейровизуализационной методики, наиболее предпочтительно МРТ;
- электроэнцефалография;
- рентгенография черепа;
- миелография, которая позволяет выявить дефекты верхнего шейного отдела спинного мозга, ствола мозга, а также локализацию мозжечка ниже черты Чемберлена в области затылочного отверстия.
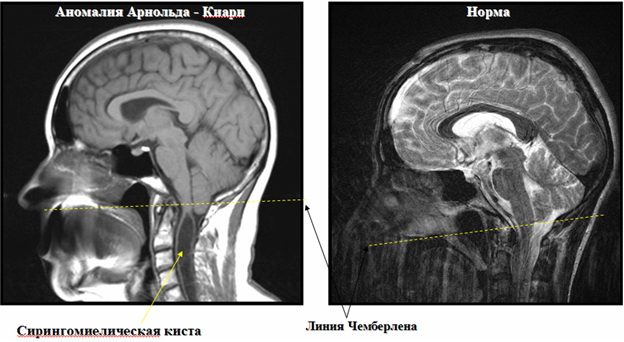
Результаты МРТ при аномалии Арнольда Киари с сирингомиелической кистой и в норме
Лечение
Тактика лечения при различных типах пороков развития мозжечка Арнольда-Киари обычно является консервативной либо продумывается нейрохирургом и представляет собой декомпрессию, наложение шунта (при выраженной гидроцефалии) или краниотомию затылочного отверстия.
Однако, благодаря докторской диссертации доктора М.Б. Ройо Сальвадора была разработана новаторская техника этиологического лечения — Filum System, которая направлена на устранение причины заболевания и патологического механизма натяжения путем хирургического минимально инвазивного рассечения концевой нити. Преимуществом методики является возможность остановить болезнь Арнольда Киари при минимальных рисках (смертность – 0%), главное выявить мальформацию и провести операцию как можно раньше. Она обычно занимает не более 45 минут и позволяет добиться симптоматического улучшения состояния, а в отдельных случаях даже поднятия миндалин мозжечка. Несмотря на короткий восстановительный период, методика имеет ряд недостатков:
- после операции остается небольшой шов;
- может возникать субъективное ощущение снижения силы конечностей;
- улучшение мозгового кровообращения вначале постоперативного периода может вызвать перепады настроения.
Но это незначительные неудобства по сравнению с минусами затылочной краниотомии, у которой:
- смертность 1-12%;
- причина заболевания не устраняется, поэтому улучшения сохраняются непродолжительный период;
- последствия оперативного вмешательства могут быть очень серьёзными и непредсказуемыми, включая отек мозга, дальнейшее опущение миндалин мозжечка, усугубление неврологической симптоматики, гемодинамические нарушения, гидроцефалию, пневмоэнцефалию, внутримозговые кровоизлияния, тетрапарез, неврологический дефицит и т.д.
Читайте также:
