
Прогрессирующая миоклоническая эпилепсия лафора

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Прогрессирующая миоклонус-эпилепсия относится к полиэтиологическим синдромам. В настоящее время выделено около 15 нозологических форм, сочетающихся с прогрессирующей миоклонус-эпилепсией. Прогрессирующей миоклонус-эпилепсией называют сложный синдром, включающий сочетание миоклонуса, эпилепсии, когнитивных нарушений и различных других неврологических нарушений (чаще всего мозжечковой атаксии) с прогрессирующим течением.
Диагностическая триада прогрессирующей миоклонус-эпилепсии:
- Миоклонические припадки.
- Тонико-клонические судорожные припадки.
- Прогрессирующие неврологические расстройства (обычно атаксия и деменция).

[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Заболевания, при которых встречается прогрессирующая миоклонус-эпилепсия
Прогрессирующая миоклонус-эпилепсия встречается при следующих заболеваниях:
Болезни, пограничные с прогрессирующей миоклонус - эпилепсией (сочетание эпилепсии и миоклонуса):
- Сочетание первичной эпилепсии и семейных миоклоний (редко)
- Болезнь Тея-Сакса (Tay-Sachs)
- Фенилкетонурия
- Липофусциноз новорожденных (синдром Santavuori-Haltia)
- Подострый склерозирующий панэнцефалит
- Болезнь Вильсона-Коновалова
- Болезнь Крейтцфельдта-Якоба
Острые состояния, при которых возможно появление миоклонус-эпилепсии:
- Интоксикация метилбромидом, висмутом, стрихнином.
- Вирусные энцефалиты.
[7], [8], [9], [10], [11], [12], [13]
Это заболевание описано в двух подгруппах больных. Одна форма выявлена впервые в Финдляндии и была названа впоследствии балтийским миоклонусом. Другая - на юге Франции (Марсель) и называется в настоящее время средиземноморским миоклонусом.
Диагностические критерии болезни Унферрихта-Лундборга включают:
- Начало болезни в возрасте между 6 и 15 годами(в 86 % случаев - между 9 и 13 годами).
- Тонико-клонические эпилептические припадки.
- Миоклонус.
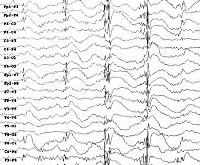
- ЭЭГ: пароксизмы спайков или комплексов полиспайк-волна с частотой 3-5 в сек.
- Прогрессирующее течение с присоединением грубой мозжечковой атаксии и деменции.
Миоклонус при болезни Унферрихта-Лундборга, как и при всех прогрессирующих миоклонус-эпилепсиях, относится к корковому миоклонусу. Он может быть как спонтанным и наблюдаться в покое, так и связанным с движениями (акционный миоклонус или миоклонус действия) и тем самым существенно затруднять повседневную активность больного. Миоклонические подёргивания провоцируются также сенсорными стимулами (стимул-сенситивный или рефлекторный миоклонус) такими как прикосновение, свет, звук и др. Миоклонус может иметь разное распределение по телу и вариирует по интенсивности даже у одного и того же больного. Обычно он асинхронный, может преобладать в одной конечности или одной половине тела, при усилении он может распространяться на другие части тела и иногда протекает в виде генерализованного миоклонического приступа без или с минимальным нарушением сознания. У большинства больных миоклонус имеет прогрессирующее течение.
У большинства пациентов развивается выраженная мозжечковая атаксия и деменция.
У больных средиземноморским миоклонусом (то, что раньше называли синдромом Рамсея Ханта) эпилептические припадки и деменция выражены весьма слабо и в отдельных случаях могут даже отсутствовать. Отвественный ген при болезни Унферрихта-Лундберга расположен на 21 хромосоме, что было подтверждено у больных со средиземноморским вариантом болезни.
Заболевание наследуется по аутосомно-рецессивному типу и начинается в возрасте 6-19 лет. Манифестным проявлением являются генерализованные тонико-клонические эпилептические припадки. Последние часто сочетаются с парциальными затылочными пароксизмами в виде простых галлюцинаций, скотом или более сложных зрительных расстройств. Зрительные пароксизмы - характерный признак болезни Лафора, наблюдаемый у 50 % больных уже на ранних стадиях заболевания. Вслед за эпилептическими приступами обычно развивается тяжёлый миоклонус покоя и действия. Атаксия нередко замаскирована тяжёлым миоклонусом. Нарушения когнитивных функций могут проявляться уже в дебюте болезни. Более грубые психические нарушения характерны для развёрнутой стадии заболевания. Возможно преходящая корковая слепота. В терминальной стадии больные прикованы к постели, у них отмечается деменция. Летальный исход наступает через 2-10 лет от начала заболевания.
Диагноз. При световой микроскопии обнаруживаются тельца Лафора в коре мозга, ткани печени и скелетных мышцах. Наиболее информативным и доступным методом является исследование биоптатов кожи, особенно в области предплечья.
[14]
Цероидный липофусциноз (церебро-ретинальные дегенерации) относится к липидозам и характеризуется отложением аутофлюоресцентных липопигментов в центральной нервной системе, гепатоцитах, сердечной мышце, сетчатке. Первичный биохимический дефект, лежащий в основе заболевания, неизвестен. Цероидный липофусциноз является одной из причин прогрессирующей миоклонус-эпилепсии. Выделяют несколько типов цероидного липофусциноза: инфантильный, поздний инфантильный, ранний ювенильный или промежуточный, ювенильный, форма взрослых.
Инфантильный тип Сантавуори-Халтиа манифестирует после 6-8 мес. и в строгом смысле не относится к прогрессирующим миоклонус-эпилепсиям.
[15], [16], [17], [18], [19], [20], [21]
[22], [C. - PubMed - NCBI" target="_blank" rel="noopener noreferrer">23]
Болезнь Гоше (Gaucher) известна в трёх формах: инфантильной (тип I), ювенильной (тип II) и хронической (тип III). Последний тип болезни Гоше может проявляться прогрессирующей миоклонус-эпилепсией. Заболевание обусловлено недостаточностью бета-глюкоцереброзидазы и характеризуется накоплением глюкоцереброзида в различных тканях организма.
[24], [25]
[26], [27], [28], [29]

Болезнь Лафоры — наследственная миоклоническая эпилепсия, при которой наблюдается отложение полисахаридных веществ в различных тканях, в первую очередь в церебральных структурах. В клинике преобладают миоклонические пароксизмы, генерализованные эпиприступы, прогрессирующая деменция, психические нарушения и расстройства зрения. Диагностика включает оценку неврологического статуса, визометрию, офтальмоскопию, ЭЭГ, томографию головного мозга, исследование биоптатов кожи. Лечение малоэффективно, представляет собой сочетание антиконвульсантной терапии с курсами тетракозактида.
МКБ-10
- Причины болезни Лафоры
- Симптомы болезни Лафоры
- Диагностика болезни Лафоры
- Лечение болезни Лафоры
- Цены на лечение
Общие сведения
Болезнь Лафоры получила свое название благодаря испанскому психоневрологу, описавшему ее в 1911 г. Наряду с клиникой заболевания Родригес Лафора дал описание специфических включений, обнаруживаемых в цитоплазме церебральных и спинальных нейронов при этой патологии. Позже включениям дали название тельца Лафоры. Они были выявлены и в других тканях организма: сердце, печени, потовых железах, скелетных мышцах.
Болезнь Лафоры — отдельный вид миоклонус-эпилепсии, который дебютирует на втором десятилетии жизни и характеризуется миоклоническими феноменами, генерализованными эпиприступами, прогрессирующим распадом когнитивных и психических функций. Неуклонно усугубляющееся течение и наследственный характер заболевания легли в основу другого его названия — прогрессирующая семейная миоклонус-эпилепсия. В неврологии болезнь Лафоры известна также под названиями болезнь телец Лафоры и миоклоническая эпилепсия II типа.
Причины болезни Лафоры
Болезнь Лафоры обусловлена наличием генной мутаций в 6-й хромосоме (локусы 6q24 и 6p22.3). У 80% пациентов аберрации затрагивают ген EPM2A, кодирующий белок лафорин, который участвует в регуляции метаболизма гликогена. В остальных случаях аберрации обнаруживаются в гене NHLRC1, отвечающем за продукцию белка малина. Болезнь Лафоры имеет аутосомно-рецессивное наследование. Если ребенок получает дефектный ген и от отца, и от матери, то со временем у него возникают нарушения углеводного обмена с отложением в тканях сходных с гликогеном амилопектин-подобных полисахаридов в виде телец Лафоры. Накопление последних носит диффузный характер, наибольшая концентрация наблюдается в таламусе, гиппокампе, зубчатом ядре мозжечка, черной субстанции.
Происходящие метаболические процессы, ведущие к образованию полисахаридных включений, пока не изучены. Известно, что небольшие запасы гликогена в нейронах являются благоприятными, поскольку повышают их устойчивость к гипоксии. Вероятно, чрезмерное накопление амилопектин-подобных полисахаридов приводит к нарушению метаболизма нейронов и их апоптозу (гибели). Результатом становятся прогрессирующие дегенеративные и атрофические изменения церебральных тканей, обуславливающие неуклонное усугубление клинической симптоматики.
Симптомы болезни Лафоры
Болезнь Лафоры манифестирует в возрастном промежутке 10-18 лет. Наиболее часто дебютом заболевания становиться генерализованный эпиприступ, в отдельных случаях — психическое расстройство. Возможно начало с миоклонических или фокальных затылочных пароксизмов. Миоклонические приступы характеризуются кратковременными внезапно возникающими асинхронными и неритмичными сокращениями отдельных мышечных групп. Могут провоцироваться внешними воздействиями (громким звуком, вспышкой света, грубым прикосновением, эмоциональным триггером и т. п.). Фокальные миоклонические пароксизмы могут быть приняты за мышечные подергивания и первоначально трактоваться как проявления невроза. Распространенные миоклонии конечностей провоцируют гиперкинезы и затрудняют активные движения; они всегда исчезают в период сна. Затылочные эпиприступы протекают с сохраненным сознанием, имеют место преходящие зрительные скотомы (возможна полная транзиторная слепота) и/или зрительные галлюцинации.
Болезнь Лафоры сопровождается когнитивным снижением, психическими отклонениями и расстройством зрительной функции. У некоторых детей сложности обучения отмечаются еще до появления первых симптомов дебюта заболевания. После начала болезни наблюдается прогрессирующая деменция. В психической сфере возможны эйфория, агрессивность, ажитация, спутанность сознания, галлюцинаторный синдром. Зрительные расстройства возникают вследствие атрофии зрительных нервов и дегенеративной ретинопатии.
С течением болезни Лафоры генерализованных тонико-клонических эпиприступов становиться меньше, но усугубляются миоклонические пароксизмы. Они становятся более частыми и двусторонними. При генерализации миоклоний наблюдается пароксизм с утратой сознания. В развернутой стадии происходит нарастание интеллектуальных расстройств, появляется и усугубляется атаксия, возникают тазовые нарушения, снижение зрения прогрессирует вплоть до амавроза.
Болезнь Лафоры длится в среднем около 10 лет. В терминальной стадии наблюдается глубокая деменция и практически постоянные миоклонии. Гибель пациентов наступает от присоединения интеркуррентных инфекций. В отдельных случаях, когда болезнь манифестирует после 20-летнего возраста, ее продолжительность может составлять 20-30 лет.
Диагностика болезни Лафоры
В неврологическом статусе определяется мышечная диффузная гипотония, выраженная атаксия (резкая неустойчивость в позе Ромберга, грубые нарушения выполнения координаторных проб), дизартрия. Пирамидные проявления мало характерны. На консультации офтальмолога при проверке остроты зрения выявляется различная степень ее снижения; офтальмоскопия диагностирует ретинопатию и/или атрофию зрительных нервов. Периметрия, выполненная в период затылочного пароксизма, может установить наличие мерцающей скотомы.
Электроэнцефалография определяет наличие синхронных медленных волн альфа-ритма наряду с кратковременными высокоамплитудными спайками (пиками). Проведение функциональных проб показывает усиление эпи-активности и возникновение миоклонических подергиваний в ответ на фото- и фоностимуляцию. КТ и МРТ головного мозга позволяют визуализировать диффузную атрофию церебральных полушарий и тканей мозжечка.
Патогномоничным диагностическим критерием выступает обнаружение телец Лафоры в протоках потовых желез. Для выявления этого критерия проводят гистологическое исследование взятых путем биопсии кожи образцов. Тельца Лафоры также могут быть выявлены при биопсии мышцы или биопсии печени. Дифференцировать болезнь Лафоры следует от других видов эпилепсии у детей, в первую очередь от юношеской миоклонической эпилепсии, от различных энцефалопатий, миоклонической мозжечковой диссинергии Ханта, наследственных обменных заболеваний (болезни Андерсена, болезни Мак-Ардля, болезни Тея-Сакса с поздним дебютом).
Лечение болезни Лафоры
Патогенетической терапии не существует. Лечение носит симптоматический характер. На первое место выходит купирование эпилептического синдрома. С этой целью эпилептологи применяют как традиционные антиконвульсанты (вальпроевая к-та), так и препараты нового поколения (леветирацетам, топирамат). Поскольку эпилепсия при болезни Лафоры с трудом поддается антиконвульсантному лечению, зачастую эти фармпрепараты комбинируют с этосуксимидом, фенобарбиталом, клоназепамом.
Параллельно с постоянной противоэпилептической терапией назначают курсы внутримышечного введения тетракозактида (от 10 до 15 инъекций на 1 курс). К сожалению, существующее лечение оказывается малоэффективным и не способно остановить прогрессирование клинической симптоматики.
Миоклоническая эпилепсия — разновидность эпилептических приступов. Характеризуется более мягким течением. Впервые болезнь проявляется у младенцев или детей раннего возраста. Дебют миоклонической эпилепсии во взрослом возрасте нетипичен. Данная форма заболевания характеризуется вялым подергиванием мышц. Симптомы напоминают тики или гиперкинезы. В некоторых случаях возможно тяжелое течение. В международной классификации болезней МКБ-10 миоклоническая эпилепсия имеет код G40.3.
Пройти диагностику и лечение заболевания в Москве можно в Юсуповской больнице. Обследование проводят опытные неврологи и эпилептологи с использованием современного медицинского оборудования. Терапия соответствует европейским стандартам качества и безопасности.

Причины миоклонической эпилепсии
В настоящее время точные причины развития миоклонической эпилепсии не установлены. Однако, врачи выделяют несколько предрасполагающих факторов. Среди них:
- Наследственная предрасположенность. Определенные виды миоклонической эпилепсии имеют наследственную обусловленность. Например, болезнь Унферрихта-Лундборга, синдром Драве. Если у одного из родственников была диагностирована эпилепсия, то последующие поколения находится в группе риска. Вероятность возникновения заболевания составляет 20-30%.
- Внутриутробная инфекция. Некоторые инфекционные агенты способны проникать через плацентарный барьер. В результате повышается риск развития не только миоклонической эпилепсии, но и психических расстройств, дефектов развития. Миоклоническая эпилепсия развивается в конце 2-3 триместра.
- Заболевания в период беременности не инфекционного характера. К данной группе болезней относится сахарный диабет, патология щитовидной железы, почечная, печеночная, сердечная недостаточность.
- Бесконтрольный прием препаратов во время беременности. Некоторые лекарственные средства обладают выраженными тератогенными свойствами. Поэтому их прием негативно сказывается на развитии плода. Доктора советуют во время беременности избегать приема лекарств. При возникновении необходимости все назначения должны быть выполнены врачом.
- Спонтанные мутации. Причины подобных изменений до сих по не изучены. Выделяют провоцирующие факторы, наличие которых способствует мутационным процессам. Среди них стресс, резкие перепады температуры, чрезмерная физическая нагрузка.
Симптомы миоклонической эпилепсии
Клиническая картина миоклонической эпилепсии зависит от вида заболевания. Среди основных патологических симптомов выделяют:
- Судороги. Заболевание характеризуется миоклоническими приступами. Они не сопровождаются выраженным болевым синдромом. Судороги чаще всего затрагивают конечности, реже лицо и туловище. В среднем продолжительность припадка составляет 10-20 минут. Сознание при этом сохраняется.
- Потеря сознания. Возникает крайне редко. Характерна для юношеской формы.
- Тонико-клонические судороги. Данная форма припадка сопровождается потерей сознания, болезненным сокращением мышц.
- Олигофрения. Умственная отсталость встречается в различных вариантах. Чаще всего это расстройство творческого мышления, интеллекта.
- Психические расстройства. Выражаются галлюцинациями, неврозами и пограничными состояниями.
Определение клинической симптоматики необходимо для подбора корректной терапии. Врачи Юсуповской больницы разрабатывают индивидуальный план лечения для каждого пациента.
Диагностика миоклонической эпилепсии
Миоклоническая эпилепсия требует проведения комплексной диагностики. Обследование включает в себя:
- Сбор жалоб и анамнеза заболевания. На первичном осмотре невролог опрашивает пациента об имеющихся жалобах, времени их появления, выраженности симптоматики. Помимо этого, уточняется продолжительность приступов, состояние после них.
- Неврологический осмотр. Врач оценивает рефлексы, проводит тестирование для оценки состояния нервной деятельности.
- ЭЭГ. Благодаря исследованию получается установить патологическую активность различных структур головного мозга. Подобным образом определяется очаг поражения.
- МРТ. Для усиления эффекта обследование проводится с использованием контраста. Миоклоническая эпилепсия не характеризуется наличием структурных изменений в головном мозге. Однако, бывают исключения.
В Юсуповской больнице для диагностики миоклонической эпилепсии используется современное медицинское оборудование. Оно позволяет быстро и эффективно установить наличие заболевания. На основании полученных данных назначается корректная терапия.
Виды миоклонической эпилепсии
Миоклоническая эпилепсия делится на несколько видов. Рассмотрим основные из них.
Диагностируется в 30-40% случаев. Характеризуется симптомами по типу гиперкинезов или тиков. Слабая степень развития заболевания может протекать незаметно на протяжении многих лет. На фоне младенческой миоклонической эпилепсии не страдает интеллектуальное развитие. Заболевание может проявиться у ребенка с 2 месяцев до 3 лет.
Выявляется на первом году жизни. Симптоматически напоминает младенческую миоклоническую эпилепсию. Синдром Драве вызывает выраженные психические расстройства. Они проявляются олигофренией, умственной отсталостью. Без корректного лечения количество приступов увеличивается до нескольких раз в неделю.
Считается генетическим заболеванием. Характеризуется тяжелой неврологической симптоматикой. Болезнь Унферрихт — Лундборга также сопровождается психическими нарушениями. Первый приступ чаще всего проявляется в период полового созревания. Болезнь диагностируется в 10-20% случаев.
Чаще диагностируется у взрослых. Миоклоническая эпилепсия юношеского возраста характеризуется тонико-клоническими пароксизмами. Болезнь не сопровождается неврологическими расстройствами, психическими нарушениями. Сознание на фоне приступа остается ясным.
Абсансы — разновидность эпилептического приступа. Считаются одним из симптомов миоклонической эпилепсии.
В зависимости от прогрессирования эпилепсии выделяют следующие формы болезни:
- Прогрессирующая. Клиническая картина эпилепсии постепенно нарастает. По мере прогрессирования заболевания увеличивается риск летальных исходов. В некоторых случаях болезнь плохо поддается лечению.
- Стабильная. Симптоматика остается на примерно одном уровне.
- Ремитирующая. Признаки эпилепсии могут медленно развиваться, а затем затухать на продолжительное время. Со временем возможно полное исчезновение патологических признаков.
Лечение миоклонической эпилепсии
Для терапии миоклонической эпилепсии применяются медикаментозные средства. Врачи Юсуповской больницы применяют следующие группы препаратов:
- противоэпилептические;
- барбитураты;
- транквилизаторы;
- ноотропы.
Каждому пациенту разрабатывается индивидуальный план лечения. Он учитывает форму миоклонической эпилепсии, стадию развития, возраст больного и наличие сопутствующих заболеваний. Такой подход позволяет быстро и эффективно подобрать терапию. В Юсуповской больнице прием ведут опытные неврологи, эпилептологи, психиатры. Реабилитацию проводят квалифицированные массажисты, инструкторы ЛФК. Для того чтобы записаться на прием, необходимо позвонить по телефону или оставить заявку на официальном сайте больницы.
Этиология. Прогрессирующие миоклонус-эпилепсии (ПМЭ) или, правильнее, - прогрессирующие формы эпилепсии с миоклонусом, относятся к наследственно – дегенеративным заболеваниям ЦНС. При многих из них верифицированы патологические гены, детерминирующие развитие заболевания. Данные формы принадлежат к симптоматической генерализованной эпилепсии и выделены в Проекте классификации 2001 года в отдельную группу.
- Первично – генерализованный миоклонус. Эпилептический миоклонус, наблюдаемый при различных синдромах идиопатической генерализованной эпилепсии. Мышечное сокращение (миоклонус) следует за пик - волновым разрядом на ЭЭГ с интервалом около 50 мсек.
- Корковый рефлекторный миоклонус. Наблюдается при фокальных формах эпилепсии; особенно характерен для эпилепсии Кожевникова. Обусловлен очаговой стимуляцией сенсомоторной коры (сенсорный стимул с периферии).
- Ретикулярный рефлекторный миоклонус. Возникает при некоторых формах генерализованной эпилепсии.
В настоящее время, во многом благодаря успехам молекулярной генетики, верифицированы и систематизированы различные формы ПЭМ, в том числе, и казуистически редкие. Установлена частота ПЭМ: 10% среди всех форм миоклонических эпилепсий и около 1% среди всех случаев эпилепсии у детей и подростков [Genton и соавт., 2002].
Классификация ПЭМ была предложена марсельской группой (Genton и соавт.) в 1993 году. Авторы выделили 3 группы ПЭМ:
I. ПЭМ с известным биохимическим дефектом.
- MERRF – синдром: миоклонус-эпилепсия с рваными красными мышечными волокнами.
- Сиалидоз: синдром миоклонуса с вишнево-красным пятном на глазном дне.
- Болезнь Гоше, тип 3.
- GM2 ганглиозидоз, юношеский тип.
II. ПЭМ с биологическими и/или патологическими маркерами, но неизвестным биохимическим дефектом.
- Цероидный липофусциноз (преимущественно, поздний инфантильный тип – болезнь Бильшовского – Янского).
- Болезнь Лафора.
- Хорея Гентингтона (детская форма).
- Синдром миоклонуса движения с почечной недостаточностью.
III. ПЭМ дегенеративного типа.
- Болезнь Унферрихта - Лундборга (балтийский миоклонус, средиземноморский миоклонус).
- Дентато-рубрально-паллидо-луизиановая атрофия.
- ПЭМ с целиакией.
Все данные синдромы имеют ряд общих клинических признаков, что значительно затрудняет установление точного диагноза. Мы обобщили симптомокомплексы, которые характерны для всех форм ПЭМ.
Необходимым условием при обследовании пациентов с подозрением на ПМЭ является проведение офтальмологического исследования, биохимии крови и мочи, молекулярно – генетического исследования; по показаниям – мышечная биопсия. При проведении МРТ головного мозга в динамике отмечается нарастающая диффузная корково – подкорковая атрофия при отсутствии локальных изменений.
Прогноз неблагоприятный, и при всех формах наступает тяжелая инвалидизация.
Терапия. В лечении прогрессирующих форм эпилепсии с миоклонусом апробированы многие АЭП без существенного эффекта или с временным улучшением. По мнению большинства авторов, вальпроаты в монотерапии и бензодиазепины в комбинации остаются препаратами выбора при длительном лечении всех форм ПЭМ [Genton & Dravet, 1996]. Положительные результаты получены при применении леветирацетама и топирамата [Genton и соавт., 2002]. Препараты группы карбамазепина и окскарбазепин противопоказаны.
Стартовая терапия осуществляется с леветирацетама . Дозировки кеппры составляют 1000-4000 мг/сут (30-60 мг/кг/сут) в 2 приема. Препарат обладает выраженным антимиоклоническим эффектом, снижает частоту генерализованных клонико – тонико – клонических приступов, блокирует фотосенситивность; хорошо переносится при длительном приеме.
Препараты второго выбора – производные вальпроевой кислоты. Применяется конвульсофин в высоких дозах 1500-3000 мг/сут (40-100 мг/кг/сут). Препарат особенно эффективен в отношении атипичных абсансов; воздействует на генерализованные судорожные приступы и эпилептический миоклонус.
Препарат третьего выбора – топирамат. Топамакс назначается в монотерапии в дозе 100-400 мг/сут (3-10 мг/кг/сут) в 2 приема. Высоко эффективен при генерализованных судорожных и фокальных приступах.
Когда монотерапия становится неэффективной, подключаются вспомогательные препараты. Наиболее оптимальна комбинация кеппры и вальпроатов. При преобладании атипичных абсансов и негативного миоклонуса, может быть эффективной комбинация кеппры или вальпроатов с суксилепом. Дозировки суксилепа составляют 500-1250 мг/сут (20-35 мг/кг/сут) в 3 приема. В развернутой стадии заболевания приходится подключать бензодиазепины. Фризиум обычно добавляется к леветирацетаму или вальпроатам в дозе 10-60 мг/сут (в среднем, около 1 мг/кг/сут) в 2-3 приема. Особенно эффективен при частых серийных приступах и эпилептическом миоклонусе.
В отдельных публикациях отмечается урежение приступов у больных ПМЭ на фоне приема примидона (гексамидин), который в России сейчас не применяется.
В последние годы убедительно доказан положительный эффект высоких доз ноотропила (пирацетам) в урежении частоты миоклонических приступов при различных формах ПЭМ. Koskiniemi и соавт. (1998), продемонстрировали достоверное уменьшение миоклонуса, улучшение моторных и когнитивных функций у 20 пациентов, страдающих болезнью Унферрихта – Лундборга, при назначении им пирацетама в дозе 24 г в сутки перорально. Genton и соавт. (2002) сообщают о положительном эффекте высоких доз пирацетама (45 г в сутки !) при болезни Унферрихта – Лундборга. Нами получены положительные результаты при внутривенном капельном введении пирацетама в дозе до 35 г в сутки при балтийском миоклонусе [Мухин К.Ю. и соавт., 2002]. Рекомендованные средние дозы ноотропила составляют около 1 мг/кг/сут (не более 40 г) при внутривенном капельном введении. Предлагается эффективная комбинация леветирацетама и пирацетама в дозе 10-15 г/сут перорально как длительная поддерживающая терапия при ПМЭ [Maggauda и соавт., 2004].
В отдельных публикациях указывается на эффективность препаратов заместительной терапии при ПЭМ. Это N - ацетилцистеин и 5-гидрокситриптофан при болезни Унферрихта – Лундборга [Selwa, 1999]; коэнзим Ку 10 и цитомаг при MERRF синдроме [П.А. Темин и соавт., 1999]; антиоксиданты при нейрональном цероидном липофусцинозе [Santavuori и соавт., 1988].
Алкоголь может быть использован с осторожностью у взрослых пациентов, так как он обладает антимиоклоническим эффектом и может снижать выраженность эпилептического миоклонуса и подкорковых гиперкинезов на несколько часов.
Прогноз при ПМЭ неблагоприятный. Все формы имеют прогредиентное течение, хотя степень нарастания клинических симптомов при всех синдромах различна. Медикаментозная терапия оказывает лишь временный эффект и является паллиативной.
Читайте также:
