
Нарушения апоптоза нервных клеток
АПОПТОЗ НЕЙРОНА — ОБЩИЙ МЕХАНИЗМ ПАТОГЕНЕЗА ПРИ ЗАБОЛЕВАНИЯХ НЕРВНОЙ СИСТЕМЫ
И. А. Завалишин, М. Н. Захарова НИИ неврологии РАМН, Москва
На современном этапе исследование патогенеза заболеваний и повреждений нервной системы осуществляется с общебиологических позиций. В результате сложилось мнение об общих механизмах формирования патологического процесса при этих состояниях. Следует отметить, что многие изученные пути поражения нервной системы являются избыточным выражением существующих в рамках нормального гомеостаза реакций, что может быть обусловлено как экзогенными, так и эндогенными причинами. Обращает на себя внимание, что общие механизмы заболеваний нервной системы могут реализоваться на разных этапах патологического процесса. Следует также отметить, что большинство из этих данных получено в экспериментальных условиях, в связи с чем перенос их на патологию человека ограничен и требует чрезвычайной осторожности.
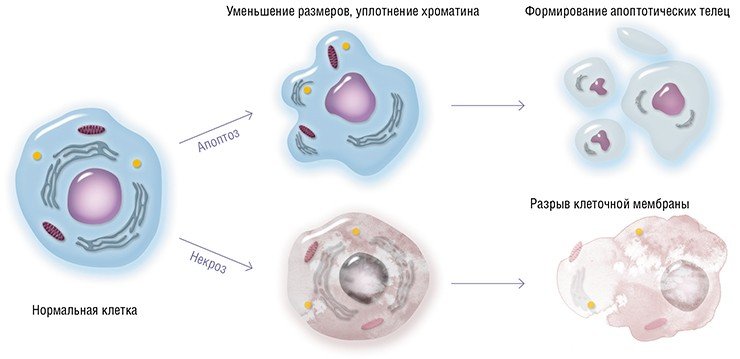
В настоящее время является общепризнанным то, что ключевой фактор патогенеза заболеваний нервной системы — гибель нейрона, может быть двух видов: программированная клеточная смерть (апоптоз) и патологическая клеточная смерть (некроз). При этом прекращение жизнедеятельности клетки в процессе апоптоза и некроза имеют четкие морфологические различия.
Примером программированной смерти нейронов служит их гибель в процессе эмбриогенеза. Все более очевидной становится роль апоптоза как при острых заболеваниях и повреждениях нервной системы (ишемия, травма), так и при нейродегенеративных болезнях (болезнь Альцгеймера, боковой амиотрофический склероз, болезнь Паркинсона).
Регуляция апоптоза в нервной системе осуществляется многочисленными сигнальными системами. Причем пути реализации этого процесса могут быть различными: модуляция активности ферментов, модуляция факторов транскрипции (р 53, АР-1, NF -кВ), прямая активация генов раннего немедленного ответа ( c - jun , c - fos ).
В настоящее время выделены три фазы апоптоза: инициации (индукции), эффекторная и деградации. В качестве инициирующих апоптоз факторов могут выступать: глутамат, (3-амилоид, депривация ростковых факторов, свободнорадикальные соединения, гипогликемия.
Первичная реакция со стороны нервной клетки на апоптотическое воздействие, по-видимому, реализуется генами раннего немедленного ответа. Активация этих генов рассматривается как один из основных, сохранившихся в эволюции, компонентов нейронального ответа на повреждение. Эти гены относятся к протоонкогенам, причем наиболее постоянно в центральной нервной системе отмечается экспрессия c - jun . Его продуктом является регуляторный протеин с- Jun , который относится к факторам транскрипции, реализующим клеточный ответ на повреждение через активацию или репрессию генов. Белок c - Jun участвует в регуляции клеточного цикла, дифференцировки, органогенеза, опухолевой трансформации, апоптоза. В последние годы установлено, что активация протоонкогена c - jun с повышенной экспрессией его продукта — протеина c - Jun
происходит при нейродегенеративных заболеваниях (болезнь Альцгеймера, боковой амиотрофический склероз). Современные авторы рассматривают c - jun как ранний маркер активации сигнальных систем при апоптозе [4]. Протеин c - Jun образует димеры с другими белками — D - Jim , c - Fos , ATF (активизирующий фактор транскрипции), в результате чего образуется АР-1 комплекс. При этом механизм активации апоптоза протоонкогенами c - jun и c - fos , а также их продуктом — фактором транскрипции АР-1, по-видимому, обусловлен либо синтезом патологических белков, либо индукцией образования гипотетического апоптотического фактора. Активизация генов немедленного ответа в нейроне может осуществляться через протеинкиназный каскад p 21 ras - MAPK или сфингомиелиназо-церамидный сигнальный путь. В результате повышается транскрипция этих генов, что способствует развитию апоптоза.
За реализацию эффекторной фазы апоптоза в любой клетке, в частности, в нейроне, ответственны так называемые каспазы. К последним у человека относится, например, интерлейкин-1р-конвертирующая протеаза ( ICE ) или каспа-за-1 [13]. В настоящее время выделены 3 класса каспаз ( ICE , CED -3 и N EDD -2/ ICH ) [9]. В норме каспазы находятся в неактивном состоянии в виде проэнзимов. Индукторы в этой ситуации выступают лишь в качестве триггеров, запуская реакции аутокатализа каспаз, т. е. самоактивации. Следует отметить, что каспазы, расщепляя как ядерные, так и цитоплазматические белковые структуры нейрона, участвуют не только в эффекторной стадии, но и в фазе деградации апоптоза, выступая в качестве основного повреждающего фактора в этом процессе.
Регуляция апоптоза во II стадии (эффекторной) осуществляется преимущественно белками семейства Вс1-2, причем выделяют два класса этих белков: тормозящие апоптоз ( Bcl -2, bc ! — xl Bcl - w , Bfl -1, Brag -1, Mcl -1, A - l ) и индуцирующие этот процесс (Вах, Bak , Bcl - Xs , Bad , Bid , Bik , Hrk ). Все белки этого семейства во многом гомологичны между собой, что позволяет им взаимодействовать между собой. Соотношение белков Вс1-2 агонистов и антагонистов апоптоза определяет способность клетки, в том числе и нейрона, отвечать на апоптотические сигналы [7].
Допускается, что антиапоптотическое действие Вс1-2 связано с нормализацией функции митохондрий, которые участвуют в реализации апоптоза [15,16]. Конкретными механизмами этого процесса являются: 1) блокирование высвобождения из митохондрий цитоохрома-Ц; 2) участие белков Вс1-2 в формировании трансмембранных митохондриальных пор, что определяет трансмембранный потенциал, а также высвобождение различных активных соединений и ионов из митохондрий; 3) возможность проникновения этих белков в липидные структуры мембран и формирование ионных каналов, что имеет значение в субклеточном распределении Са 2+ между ядром, митохондриями и эндоплазматическим ретикулумом.
Гены семейства Вс1-2 и каспаз экспрессируются нейронами как в онтогенезе, так и в зрелой нервной системе. Опыты Martinou J. С . et al. (1994) показали, что у трансгенных мышей с избыточной экспрессией Вс1-2 мотонейроны устойчивы к апоптозу. Однако при неонатальной аксонотомии они значительно атрофируются, но выживают [8]. Повышенная экспрессия Вах выявлена при боковом амиотрофическом склерозе и болезни Альцгеймера. Вс1-2 оказывает выраженное влияние на выживание любых нейронов и, в частности, мотонейронов.
Исследование Вс1-2 иммунохимическими методами в нейронах гиппокампа при болезни Альцгеймера в зависимости от степени тяжести, клинических симптомов и нейропатологических изменений (аутопсийные исследования) показало, что в целом экспрессия Вс1-2 в нейронах нарастала по мере прогрессирования и тяжести заболевания. Однако в нейронах, в которых идентифицированы нейрофибриллярные изменения, отмечено снижение Вс1-2, то есть синтез Вс1-2 резко снижается в этих дегенерирующих нейронах. Повышение Вс1-2 выявлено в астроцитах и эндотелии сосудов при болезни Альцгеймера. Повышение Вс1-2 рассматривается авторами как защитный механизм, тормозящий апоптоз в сохранных нейронах [12].
Выявлены отличия антиапоптотического действия Вс1-2 от эффектов фактора роста нервов ( nerve growth factor , NGF ): 1) NGF вызывает морфологическую дифференцировку клеток, а Вс1-2 нет; 2) период выживания клеток под действием Вс1-2 короче, чем соответственно с NGF . Однако не продемонстрирована экспрессия Вс1-2 в ответ на NGF . Вместе с тем, Вс1-2 не подавляет апоптоз, вызванный дефицитом цилиарного нейротрофического фактора ( ciliary neurotrophic factor , CNTF ) в отличие от NGF . Это предполагает существование различных механизмов апоптоза.
Недавние исследования выявили частичную делецию гена, ответственного за экспрессию белка, ингибирующего нейрональный апоптоз, при спинальной мышечной атрофии ( NIAP , neuronal inhibitory apoptosis protein ) [13]. Этот белок гомологичен белку IAP вирусного происхождения ( baculo virus ). Установлено двойное действие полиовируса: индуцирование апоптоза за счет блока макромолекулярного синтеза, а при определенных условиях, наоборот, — проявление антиапоптотической активности. В связи с этим следует отметить, что существует гипотеза в отношении фрагментации ДНК, сопровождающей апоптоз, которая, возможно, возникла как механизм противовирусной защиты чтобы не допустить репликации вируса в клетке. В настоящее время показано, что ряд вирусных белков тормозит апоптоз в нейроне.
В 1993 г. был идентифицирован новый ген, индуцирующий апоптоз исключительно в нервной системе — это ген низкоаффинного рецептора к фактору роста нервов ( pTSNGFR ) [11].
Следует отметить, что в нейронах зрелой нервной ткани нет экспрессии Р15 NGFR , который относится к семейству, включающему и гены рецепторов к Факторам некроза опухоли, однако при болезни Альцгеймера и боковом амиотрофическом склерозе выявлена его повышенная экспрессия, соответственно, в базальных холинергических нейронах и в мотонейронах спинного мозга. Пред полагается, что повышенная экспрессия p 75 NGFR способствует образованию арахидоновой кислоты, активации перекисного окисления липидов и развитию окислительного стресса. [2]
Большое значение в развитии апоптоза отводится цитозольному фактору транскрипции ( NF -кВ), который регулирует экспрессию генов, кодирующих белки, которые участвуют в формировании иммунного ответа и реакций воспаления. NF -кВ существует в двух формах: индуцибельный (в цитоплазме и синапсах) и конститутивной (в ядре). Этот фактор выявлен в синапсах коры больших полушарий, мозжечка и гиппокампа. Установлена возможность ретроградного транспорта NF -кВ из синапса в ядро. Это новая сигнальная система для ядра. Экспрессия NF -кВ имеет важное значение в нейрональной пластичности и синаптической активности [10].
Накапливаются данные в пользу участия NF -кВ в развитии болезни Альцгеймера: (3-амилоид активирует NF -кВ через образование активных метаболитов 02, tau -белки также активируют NF -кВ. При этом NF -кВ активируется вокруг бляшек на самых ранних стадиях болезни путем взаимодействия с RAGE -рецептором, общим для tau и А (3. В свою очередь активированный NF -кВ совместно с метаболитами tau индуцирует экспрессию гена-предшественника амилоидного пептида. Вместе с тем следует отметить, что наряду с апоптотическим эффектом NF -кВ при определенных условиях может оказывать и нейропротективное действие [3].
Некроз клетки — тип клеточной смерти, принципиально отличный от упорядоченного прекращения жизнедеятельности в процессе апоптоза развивающихся нейронов. Причиной этого процесса могут стать различные патогенные факторы: гипоксия, токсемия, гипертермия и др. При некрозе наблюдаются вакуолизация, резкое набухание клеток, завершающееся лизисом.
В последние годы установлено, что гены, имеющие значение в механизмах развития апоптоза, участвуют и при формировании нейронального некроза. Так, показано, что Вс1-2 ингибирует некроз [2]. Предполагается, что этот ген регулирует внутриклеточные процессы, в одних случаях приводящие к апоптозу, в других — к некрозу. Другой антиапоптотический ген Bcl - X L подавляет не только апоптоз, но и некротическую гибель нейронов при гипоксии. Ингибиторы ICE протеаз также способны затормозить развитие не только апоптоза, но и некоторые формы некроза. Это предполагает наличие общих механизмов гибели клетки как при апоптозе, так и некрозе [14].
В нейрональной культуре повышенная экспрессия p 75 NGFR вызывается введением (3-амилоидного пептида ((3-АР), морфологически при этом наблюдается картина в большей степени похожая на некроз, чем на апоптоз. Допускается, что этот механизм может быть одним из звеньев патогенеза болезни Альцгеймера.
Выраженной нейрональной токсичностью обладают и некоторые амилоидогенные пептиды, в частности, (3-АР 1-40 и (3-АР 25-35, а также пептид прионного белка Р^Р 106-126. От концентрации, например, пептидов (3-АР зависит механизм гибели клетки — некроз или апоптоз, pr ? 106-126 индуцирует апоптоз. Сложилось мнение, что различные факторы, приводящие клетку к гибели, вызывают либо некроз (большие концентрации за короткое время), либо апоптоз (малые дозы за длительный период) [2, 14]. К этим факторам относятся активные метаболиты кислорода, концентрация внутриклеточного Са 2+ , нарушение формирования Са 2 +-каналов и Са 2+ гомеостаза, повышение чувствительности к глутамату его рецепторов, блок тахикининовых рецепторов и т. д.
Все эти исследования убеждают в существовании сложной системы регуляции апоптоза и некроза. Предполагается наличие и других пока не идентифицированных генов, регулирующих эти процессы в нервной системе.
Современный уровень знаний о молекулярных механизмах гибели нейрона при болезнях Альцгеймера, Паркинсона и Гентингтона, боковом амиотрофическом склерозе, эпилепсии, ишемии и гипогликемии явно недостаточен для понимания всех аспектов их патогенеза. Тем не менее, представляется весьма вероятным, что в повреждении нервных клеток при этих различных по этиологии заболеваниях принимают участие два стандартных механизма — окислительный стресс и эксайтотоксичность, которые могут индуцировать развитие некроза или апоптоза нейрона [1].
Таким образом, механизмы гибели нервной клетки при нейродегенеративных заболеваниях осуществляются, главным образом, по механизму апоптоза; при острых заболеваниях и повреждениях нервной системы в основном по пути некроза. Реализация этих эффектов связана с изменением экспрессии ряда онкогенов в связи с развитием реакций окислительного стресса и эксайтотоксичности, являющихся одним из общих механизмов повреждения нервной системы при различных патологических состояниях.
1. Завалишин И. А., Захарова М. Н. Оксидантный стресс — общий механизм повреждения при заболеваниях центральной нервной системы // Ж. Неврологии и психиатрии им. С. С. Корсакова. 1996, № 2, с . 111-114.
2. Bredesen D. E. Neuronal apoptosis: genetic and biochemical modulatio n. //In. Apoptosis II: The molecular basis of apoptosis in disease. Ed Tomei L. D., Cope F. 0. 1994. Cold Spring Harbor Lab. Press p. 397-421.
3. Cebollos-Picot I. The role ofoxidative stress in Neuronal Death. 1997. Springer. 203 P.
4. Herdegen F., Skene P, Bauhr M. The c-Jun transcription factor-bipotential mediator ofneuronal death, survival and regeneration// TINS, 1997. v. 20, p. 227-231.
5. Holtzman D. M., Deshmukh M. Caspases: a treatement target for neurodegenerative disease.//Nature Medicine 1997, v. 3, p. 954-955.
6. Kim T-W, Warren H. P, Jung Y-K. Alternative cleavage of Alsheimer-associated Presenilins during apoptosis by a caspase — 3 family protease.//Science 1997, v. 277, p. 373-376.
7. Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis.// Nature Medicine 1997, v. 3, p. 614-620.
8. Martinou J. K-., Dubois-Dauphin V., Staple J. K. Overexepression of bcl-2 in transgenic
mice protects neurons from naturally occurring cell death and experimental ishemia.// Neuron. 1994, v. 13, P. 1017-1030.
9 McCarthy N. J., Whyte M. K., Gilbert C. S. Inhibition of ced-3/ICE related proteases does not prevent cell death induced by oncogenes, DNA damage or the Bcl-2 Homologue Bak//J. Cell. 1997, v. 36 p. 215-227.
10. ONeill L. A. J., Kaltschmidt C. NF- кВ : a crucial transcription factor for glial and neuronal cell function.//TINS. 1997, v. 20 p. 252-258.
11- Rabizadeh S., Ohj., Zhong 1. et al. Induction ofapoptosis by the low-affinity NGF receptor.//Science 1993, v.,261, p. 345-348.
12. Satou Т ., Cummung s B. J., Cotman C. W. Immunoreactivity for Bcl-2 protein within neurons in the Alzheimers disease brain increases with disease severity. // Brain. Res. 1995, v.697, p. 35-43.
13. Schwartz L. M., Milligan С . Е . Cold thoughts of death: The role of ICE prote ases in neuronal cell death. //TINS 1996 v. 19, p. 555-562.
14. Shimizu S., Eguchi Y., Kamiike W. et al./ Retardation of chemical hypoxia-induced necrotic cell common mediators in apoptotic and necrotic sig nal transductions.// Oncogene. 1996, v. 12, p. 2045-2050.
15. Yang J., LinX., BhallaK. etal. Prevention of apoptosis by Bel-2: release of cytochrome С from mitochondria-bio blocked. //Science 1997, v. 275, p. 1129-1132.
16. ZamzamiN., SusinS., MacchettiP. Mitochondrial control of nuclear apoptosis. //J. Exp. Med.1996, v.183, p. 1533-1544.
Патология, связанная с ослаблением апоптоза
Вирусные заболевания
В норме инфицированные клетки погибают в результате активации рецептор-зависимого апоптоза, дабы предотвратить распространение вируса. Однако некоторые вирусы способны нарушать нормальную регуляцию механизма программируемой клеточной гибели, или даже активно предотвращать апоптоз. Вирусная блокада клеточной гибели может быть основана на синтезе IAP, гомологов белка Bcl-2, а также других ингибиторов апоптоза.[73]
Бакуловирусы насекомых блокируют апоптоз за счёт экспрессии IAP, блокирующих инициаторную и эффекторные каспазы. К тому же бакуловирусы экспрессируют белок p35, который связывает и ингибирует активные каспазы. В результате жизнеспособность заражённой клетки поддерживается до образования достаточного количества вирусных частиц.[74]
Вирусы позвоночных способны блокировать апоптоз путём синтеза антиапоптотических белков семейства Bcl-2, например E1B19K и белок BHRF вируса Эпштейна — Барр. К тому же вирусы позвоночных часто функционируют, предотвращая апоптоз, запускаемый клетками иммунной системы. Например, вирус ветряной оспы продуцирует серпины, блокирующие гранзим B и каспазу-8. Тем самым, инфицированная клетка избегает воздействия со стороны цитотоксических лимфоцитов, а также избегает апоптоза. Другой пример: вирус герпеса продуцирует белок v-FLIP, который блокирует апоптоз, реализуемый с участием рецепторов клеточной гибели.[74]
Опухолевые заболевания
Вторую группу заболеваний, связанных с ослаблением апоптоза, составляют злокачественные опухоли. В качестве основной причины данной патологии рассматривают соматические мутации гена, кодирующего белок p53. Порядка 50 %[74] (70 %[75]) трансформированных клеток экспрессируют мутантную форму p53. Механизм подавления программируемой клеточной гибели также может быть связан с повышенной экспрессией или мутацией гена Bcl-2.[76] Например, установлен факт рекомбинации гена Bcl-2 при лимфоме Беркитта и некоторых формах фолликулярных лимфом.[75]
Аутоимунные заболевания
Основным признаком аутоиммунной патологии является иммунная реакция против собственных клеток и тканей организма, причиной чему может быть сбой в программе негативной селекции T-лимфоцитов. Нарушение T-клеточного апоптоза позволяет выжить аутореактивным клонам T-лимфоцитов. Вдобавок нарушается формирование полного состава апоптозных аутоантигенов (свойственных организму белков-участников апоптоза), к которым должна развиться толерантность. Как следствие, малые по интенсивности проапоптозные воздействия приводят к повышению уровня аутоантигенов, участвующих в апоптозе, что в свою очередь влечёт за собой проявление клинических признаков аутоиммунной патологии. Примером могут служить аутоиммунные дерматиты, прогрессирующие при воздействии солнечных лучей или при снижении температуры окружающей среды.[76]
Патология, связанная с усилением апоптоза
Одной из групп заболеваний, связанных с усилением апоптоза, являются патологии системы крови. Чаще всего патологические процессы развиваются в результате гибели посредством апоптоза костномозговых клеток-предшественников. Причиной их гибели является недостаточность факторов выживания. Данный тип патологии приводит к развитию апластической анемии; анемии при дефиците железа, фолатов, витамина B12; талассемии; тромбоцитопении; лимфопении; нейтропении; панцитопении. Повышенная готовность к развитию апоптоза Т-лимфоцитов обнаружена при мультицентрической болезни Кастелмана.[72]
Прогрессия некоторых инфекционных заболеваний может быть связана не только с подавлением, но и наоборот, с усилением апоптоза. Индукторами программируемой клеточной гибели при этом служат бактериальные эндо- и экзотоксины. Массовый апоптоз развивается при сепсисе. Гибель лимфоцитов путём апоптоза находится в положительной корреляции с быстрой прогрессией СПИДа.[72]
Отдельную группу патологии составляют заболевания нервной системы, обусловленные атрофией определённых участков нервной ткани в результате апоптоза. Примерами таких заболеваний могут служить боковой амиотрофический склероз, болезнь Альцгеймера, спинальная мышечная атрофия и др.[72]
Апоптоз является преобладающей формой гибели миоцитов в ранний период развития инфаркта.[72] На основе экспериментальных данных было выявлено, что программируемая гибель кардиомиоцитов может быть обусловлена гипоксией, ишемией, перегрузкой клетки кальцием, воспалением, токсинами.[77] В процессе токсического (в том числе и алкогольного) гепатита основная роль также отводится апоптозу.[72]
Ряд патологических процессов, обусловленных усилением апоптоза, индуцируется внешними апоптогенными факторами. Апоптоз прогрессирует под воздействием ионизирующей радиации. При этом преимущественно гибнут лимфоидные клетки и развивается иммунная недостаточность. Аналогичный эффект дают многие химиотерапевтические препараты, используемые при лечении опухолей, а также гормоны, применяемые при лечении различных заболеваний.[72]
Введение
- Предмет и задачи патофизиологии
- История развития патофизиологии
Общая патофизиология
- Общие вопросы патофизиологии
- Региональные типовые патологические процессы
- Типовые нарушения обмена веществ
- Иммунопатология
- Патофизиология тканевого роста
- Патофизиология экстремальных состояний
- Хронопатология
Частная патофизиология
- Патология крови и кроветворения
- Патология сердечнососудистой системы
- Патология системы дыхания
- Патология системы пищеварения
- Патология выделительной системы
- Патология эндокринной системы
- Патология нервной системы
- Инфекционный процесс
Вход в систему
Поделиться
- Общие вопросы патофизиологии
- Общая нозология
- Понятие о здоровье
- Понятие о болезни
- Патогенез
- Патологическая доминанта
- Формирование порочных кругов
- Нарушение последовательности информационного процесса, определяющего развитие физиологических и патологических реакций
- Саногенез
- Первичные саногенетические механизмы
- Вторичные саногенетические механизмы
- Саногенетическая роль патогенетических механизмов
- Патогенетическая роль саногенетических механизмов
- Периоды болезни
- Болезнь как патология информационного процесса
- Нарушения информации, ведущие к развитию тех или иных патологических процессов
- Нарушения ввода (восприятия) информации
- Нарушение трансляции информации
- Патология накопления, считывания и обработки информации
- Патология реализации информации
- Программные команды как аналоги механизмов некоторых патофизиологических реакций
- Нарушения информации, ведущие к развитию тех или иных патологических процессов
- Реактивность организма
- Видовая реактивность организма
- Половая реактивность организма
- Возрастная реактивность организма
- Внешние признаки старения
- Изменения в организме связанные со старением
- Причины старения
- Конституциональная реактивность
- Индивидуальная реактивность организма
- Патофизиология клетки
- Патология некоторых составных элементов клетки
- Патология клеточных мембран
- Функции и строение биомембран
- Патология клеточного ядра
- Патология митохондрий
- Патология лизосом
- Патология эндоплазматического ретикулума
- Патология клеточных мембран
- Апоптоз
- История исследования апоптоза
- Отличия апоптоза от некроза
- Проявления апоптоза
- Генетический контроль клеточной гибели
- Заболевания, связанные с нарушением апоптоза
- Опухоли
- Аутоиммунные заболевания
- Апоптоз и вирусные инфекции
- Нарушения биоритмов клетки
- Нарушения гуморальной и нервной регуляции клетки
- Патология клетки и болезнь
- Болезни накопления
- Злокачественные опухоли
- Атеросклероз
- Гангрена
- Клетка как система
- Патология некоторых составных элементов клетки
- Общая нозология
Заболевания, связанные с нарушением апоптоза
Апоптоз лежит в основе патогенеза многих заболеваний, которые можно разделить на две группы.
Заболевания, связанные с ингибированием апоптоза.
2. Аутоиммунные болезни.
3. Вирусные инфекции (герпес, аденовирусы).
4. Заболевания, протекающие с гиперэозинофильным синдромом.
5. Нейропролиферативные заболевания (шизофрения).
Заболевания, связанные с усилением апоптоза.
2. Нейродегенеративные заболевания (болезнь Альцгеймера, паркинсонизм, боковой амиотрофический склероз, атрофия мышц спины).
3. Болезни крови (миелодиспластический синдром, апластическая анемия).
4. Ишемические повреждения (инфаркт миокарда, инсульт, реперфузионные повреждения).
5. Токсические повреждения печени.
6. Заболевания почек.
Опухоли
Увеличение количества клеток того или иного вида при различных заболеваниях может быть связано как с увеличением пролиферации, так и со способностью клеток запускать программу клеточной гибели при действии факторов, вызывающих апоптоз в норме. По-видимому, кроме усиления размножения клеток, нарушение апоптоза является одним из механизмов развития опухолевого процесса. Исследования последних лет позволили выделить некоторые механизмы увеличения резистентности опухолевых клеток к факторам, индуцирующим апоптоз. Был идентифицирован ген bcl-2. содержание которого значительно увеличивается при фолликулярной лимфоме у человека. Этот ген не способен ни ускорять прохождение клеточного цикла, ни усиливать пролиферацию. Пониженная экспрессия данного гена обнаружена в клетках, в которых апоптоз активирован. Повышенное содержание гена bcl-2 в опухолевых клеточных линиях наблюдается в случае их резистентности к действию химиотерапевтических препаратов. Введение генов, ингибирующих bcl-2, может усилить апоптоз в опухолевых клетках, что характеризует ген bcl-2, как фактор блокады апоптоза.
Важную роль в нормальной регуляции процессов апоптоза играет ген р53. Опухоли, содержащие нормальный ген р53, поддаются химиотерапии и имеют хороший прогноз. Таким образом, апоптоз при опухолях можно рассматривать как механизм защиты организма от клеток, имеющих генетические изменения, приводящие к усиленной клеточной пролиферации.
В настоящее время активно разрабатываются вопросы, являющиеся теоретической предпосылкой разработки нового направления терапии опухолевых заболеваний, связанных с угнетением генов-репрессоров и активацией генов, стимулирующих апоптоз.
Аутоиммунные заболевания
Регулирование клеточной гибели в физиологических условиях необходимо для устранения аутореактивных лимфоцитов в процессе развития и удаления избытка клеток после завершения иммунного ответа.
Аутоиммунные заболевания (аутоаллергические) являются патологическими состояниями, при которых иммунная реакция организма направлена против собственных клеток и тканей, независимо от природы фактора, вызвавшего эту реакцию.
На основании имеющихся в настоящее время данных роль апоптоза в патогенезе аутоиммунных заболеваний может быть представлена следующим образом. Активация клеточной гибели ответственна за изменение негативного отбора реактивных Т-лимфоцитов в тимусе. Нарушение Т-клеточного апоптоза в тимусе не только облегчает персистирование аутореактивных клонов, но и блокирует образование полного состава апоптозных аутоантигенов, к которым должна развиваться толерантность. При этом действие фактора, вызывающего апоптоз, даже при малой интенсивности его воздействия (например, при воздействии солнечных лучей или при снижении температуры окружающей среды) приводит к тому, что даже низкий уровень антигенов на фоне сенсибилизированной иммунной системы вызывает проявление клинических признаков аутоиммунной патологии (в данном случае, аутоиммунных дерматитов).
Апоптоз и вирусные инфекции
Гибель инфицированных вирусами клеток может рассматриваться как защитный механизм, предотвращающий распространение вирусов в организме. Инициирование апоптоза в этом случае связано с активированием специальных клеточных рецепторов на поверхности клеток-мишеней (так называемых Fas-рецепторов). Однако попадание в клетку некоторых вирусов, в частности, вируса герпеса, приводит к нарушению нормальной регуляции механизма апоптоза. Некоторые вирусы способны активно предотвращать апоптоз инфицированных клеток, используя гомолог гена bcl-2, содержащегося в геноме вируса, в то время как другие могут активно индуцировать экспрессию нативного bcl-2. Так, введение белков аденовируса может непосредственно блокировать апоптоз.
Предотвращение апоптоза играет важную роль в механизмах латентной вирусной инфекции, что зависит от экспрессии гена вируса в клетку и подавления процессов гибели поврежденной вирусом клетки.
Все клетки многоклеточных существ несут в себе потенциальную способность к апоптозу, так же как японские самураи всю жизнь носят с собой меч. И если по каким-то причинам тонкий механизм апоптоза разлаживается, последствия для организма могут оказаться самыми катастрофическими. Например, раковые клетки, блокируя систему апоптоза, приобретают бессмертие. Поэтому изучение механизмов клеточной самоликвидации является важнейшим направлением современных биомедицинских исследований: раскрытие тайн апоптоза поможет в разработке новых лекарств для борьбы с самыми тяжелыми и трудноизлечимыми болезнями современности
Каждый день и каждый час в нашем организме погибают миллионы клеток. Отшелушиваются ороговевшие клетки покровного эпителия, быстро изнашиваются и гибнут клетки слизистой ткани, выстилающей пищеварительный тракт, лейкоциты – белые клетки крови, находят свою смерть в борьбе с патогенами… Но как наше тело избавляется от специализированных клеток, когда в результате накопившихся внутренних повреждений они становятся неспособными выполнять свои функции? Одним из самых парадоксальных и удивительных механизмов, контролирующих жизнеспособность многоклеточного организма, является апоптоз – клеточная самоликвидация.
Регулярная, генетически запрограммированная гибель отдельных клеток необходима для нормального функционирования организма в целом. Все клетки многоклеточных существ обладают аппаратом апоптоза, так же как японские самураи всю жизнь носят с собой меч. Однако у этого естественного процесса есть и обратная сторона: если по каким-то причинам тонкий механизм апоптоза разлаживается, последствия для организма могут оказаться самыми катастрофическими.
Нарушения в запуске апоптоза ведут к возникновению ряда серьезных заболеваний, в том числе аутоиммунных и онкологических. Например, раковые клетки, блокируя систему апоптоза, приобретают бессмертие. Поэтому изучение механизмов клеточной самоликвидации является важнейшим направлением современных биомедицинских исследований: раскрытие тайн апоптоза поможет в разработке новых лекарств для борьбы с самыми тяжелыми и трудноизлечимыми болезнями современности.
Ферменты-киллеры
В то же время каспазы активируют ряд белков, которые участвуют в выполнении программы самоликвидации. Например, белка, который разрезает ДНК на большие фрагменты, – этот процесс, после которого целостность ДНК необратимо уничтожается, является характерной чертой апоптоза.
Сигнал на запуск
Но каким же образом клетка узнает, что ей пора самоликвидироваться? Кто и как дает указания киллерам-каспазам?
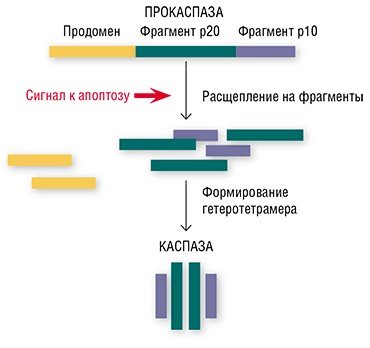
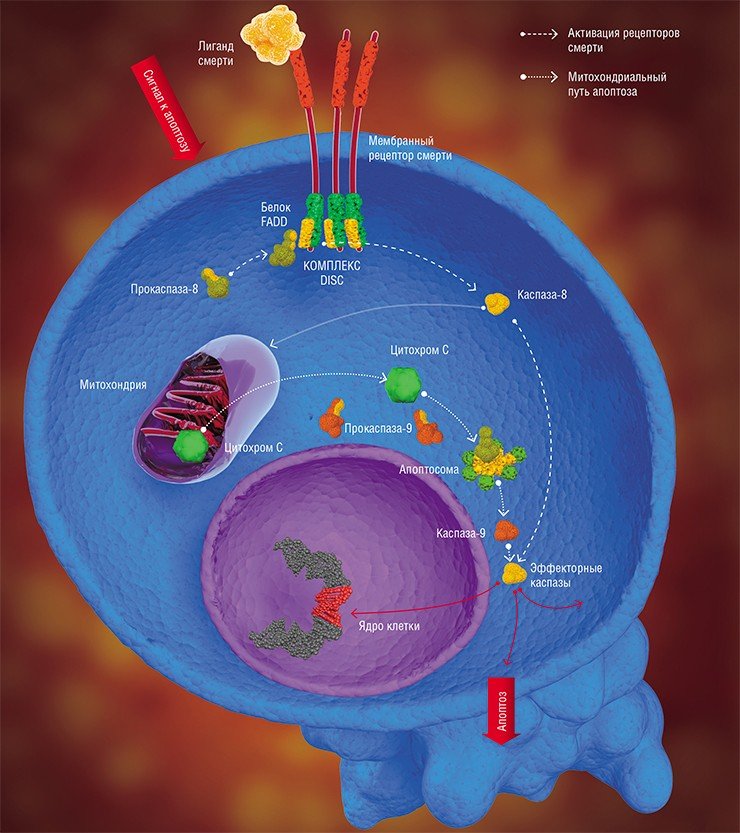
Имеется два основных пути, по которым передаются апоптопические сигналы в виде клеточных регуляторов, таких как гормоны, антигены, моноклональные антитела и другие молекулы. Это митохондриальный (или внутренний) путь, а также через особые трансмембранные белки – так называемые рецепторы смерти (DR, от англ. death receptor). В обоих случаях для запуска апоптоза должны образоваться особые инициаторные апоптотические комплексы. Затем происходит активация так называемых инициаторных каспаз, которые, в свою очередь, активируют эффекторные (разрушающие клеточные структуры) каспазы, о которых упоминалось выше (Nicholson, 1999).
Речь идет об очень интересном явлении – самоактивации прокаспазы. Такое может произойти лишь в том случае, когда две молекулы этого белка, ориентированные определенным образом относительно друг друга, образуют димер. Именно такие уникальные пространственные условия, необходимые для димеризации и каталитической активации фермента, и предоставляет прокаспазе-9 апоптосома. Образовавшаяся в результате активная каспаза-9 расщепляет эффекторные каспазы (каспазу-3 и каспазу-7), а дальше все происходит по стандартной схеме апоптоза (Green et al., 2004).
В случае рецептор-зависимого сигнального пути инициация апоптоза начинается с другого белкового комплекса, который образуется непосредственно на самом рецепторе смерти (Krammer et al., 2007; Lavrik et al., 2005).
Жить или не жить?
Итак, в отличие от голливудского детектива, в истории про апоптоз нет главного действующего лица: своевременное уничтожение поврежденных клеток и в итоге – жизнеспособность организма зависит от слаженной цепочки событий, в которой участвует множество различных белковых молекул.
И здесь очень важны количественные показатели, такие как концентрация. Сегодня изучением того, как влияет на инициацию и дальнейший ход апоптоза уровень содержания в клетке различных молекул, занимается одна из передовых областей современной науки – системная биология (Bentele et al., 2004). Основной ее постулат заключается в том, что протекание сложных процессов внутри клетки можно понять, лишь учитывая максимально большое число клеточных параметров. Для этого на основе экспериментальных данных создается компьютерная модель, которая учитывает действие множества факторов. Полученные таким образом предсказания о ходе основных клеточных процессов могут использоваться в борьбе с препятствиями человечества на пути к долгой и здоровой жизни.
Lavrik I. N., Golks A., Krammer P. H. Caspases: Pharmacological manipulation of cell death // J. Clin. Invest. 2005. V. 115, N 10. P. 2665—2672.
Krammer P. H., Arnold R., Lavrik I. N. Life and death in peripheral T cells // Nat. Rev. Immunol. 2007. V. 7. P. 532—542.
Green D. R. and Kroemer G. The pathophysiology of mitochondrial cell death // Science. 2004. V. 305. P. 626—629.
Читайте также:
